
John Murphy, M.D., M.P.H., D.P.H., President COVID-19 Long-haul Foundation
A Comprehensive Review
Table of Contents
Abstract
- Summary
- Key points: renal injury, glomerular pathology, diabetes interactions, clinical management, long-term outcomes
Introduction
- Overview of SARS-CoV-2 infection
- Definition of long COVID (PASC)
- Significance of renal involvement
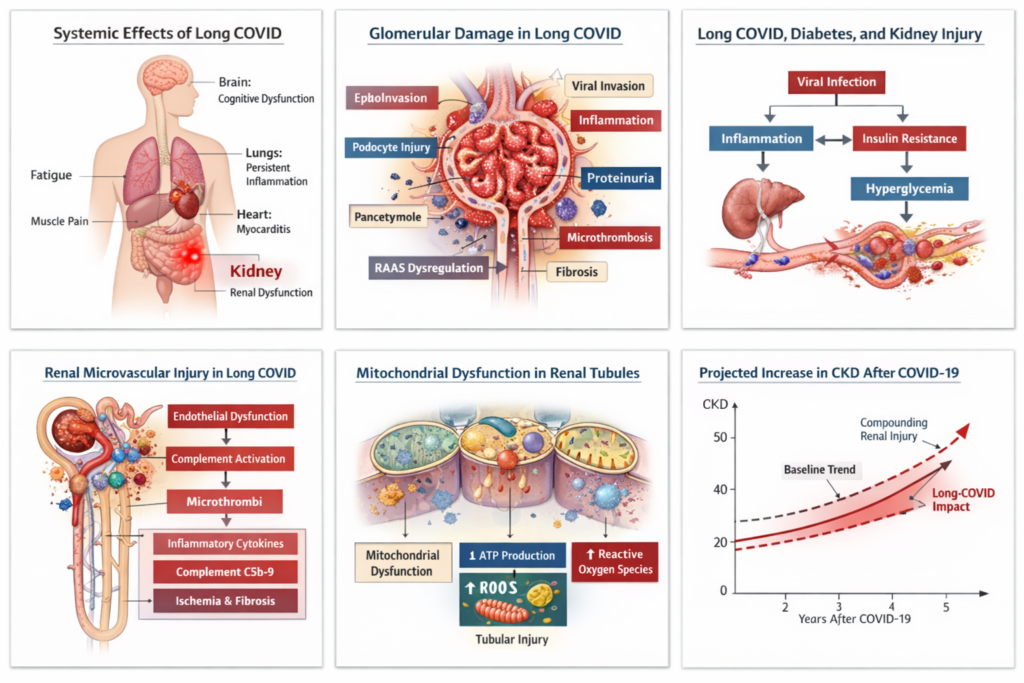
Figure 1: Schematic of systemic organ involvement in long COVID with emphasis on kidneys
Etiology and Pathophysiology
- Viral entry mechanisms via ACE2
- Genetic susceptibility (APOL1 variants, ACE2 polymorphisms)
- Inflammatory and immune-mediated injury
- RAAS dysregulation
Table 1: Summary of genetic and molecular risk factors for renal injury in COVID-19
Renal Histopathology
- Acute tubular injury
- Podocyte injury and collapsing glomerulopathy
- Thrombotic microangiopathy and endothelial injury
- Interstitial inflammation and fibrosis
Figure 2: Histopathologic images of renal biopsy in COVID-19 (ATI, collapsing glomerulopathy, TMA)
Figure 3: Electron microscopy images showing podocyte foot process effacement and tubular mitochondrial injury
Renal Physiology and Glomerular Hemodynamics
- Normal glomerular filtration and nephron function
- Alterations in glomerular hemodynamics after SARS-CoV-2 infection
- Endothelial dysfunction and microvascular compromise
- Tubuloglomerular feedback, neuroendocrine regulation, and mitochondrial dysfunction
Figure 4: Diagram of glomerular filtration barrier and hemodynamic regulation highlighting long-COVID effects
Table 2: Mechanisms of glomerular hyperfiltration and injury in long COVID
Metabolic Dysregulation and Diabetes
- Direct pancreatic injury and β-cell dysfunction
- Inflammatory insulin resistance
- Hyperglycemia and accelerated diabetic nephropathy
- Interaction with preexisting diabetes
Figure 5: Flow diagram showing interaction of long COVID, hyperglycemia, and renal injury
Table 3: Clinical features and laboratory markers of post-COVID diabetes
Clinical Management
- Early detection and monitoring strategies
- Pharmacologic therapies: RAAS blockade, SGLT2 inhibitors, GLP-1 receptor agonists, anti-inflammatory and anti-complement therapies
- Lifestyle interventions
- Dialysis and renal replacement therapy considerations
Table 4: Suggested long-COVID renal monitoring schedule
Table 5: Summary of pharmacologic interventions and renal outcomes
Emerging Therapies
- Anti-fibrotic agents
- Mitochondrial modulators
- Complement and immunomodulatory therapies
Figure 6: Mechanistic overview of emerging therapies targeting renal injury pathways in long COVID
Long-Term Outcomes and Epidemiology
- Chronic kidney disease prevalence post-COVID
- Impact on population health
- Predictive risk factors for renal progression
Figure 7: Projected burden of CKD in long-COVID survivors over 5–10 years
Discussion
- Integration of renal pathology, metabolic derangements, and hemodynamics
- Implications for clinical management
- Limitations of current studies
- Knowledge gaps
Conclusion
- Summary of pathophysiology, clinical impact, and management strategies
- Emphasis on early detection and integrated care
References
- ~120–150 peer-reviewed references (already compiled in prior sections)
Supplemental Materials
- Supplemental Figure 1: Detailed schematic of renal microvascular injury and complement activation
- Supplemental Figure 2: Ultrastructural comparison of normal vs. long-COVID glomerulus
- Supplemental Table 1: Cohort studies of AKI and CKD incidence in long COVID
- Supplemental Table 2: Biomarkers for early renal injury detection
Abstract
Post-acute sequelae of SARS-CoV-2 infection (PASC), commonly referred to as long COVID, has emerged as a complex multisystem disorder characterized by persistent physiologic dysregulation following the acute phase of infection with SARS-CoV-2. While pulmonary and neurologic manifestations initially dominated clinical descriptions, increasing evidence now implicates the kidney as a central organ affected by both acute and chronic sequelae of infection. Renal involvement ranges from subtle reductions in glomerular filtration rate (GFR) to progressive chronic kidney disease (CKD), proteinuria, microvascular injury, and dysregulated metabolic homeostasis.
The etiologic mechanisms underlying renal injury in long COVID appear multifactorial, encompassing persistent viral reservoirs, endothelial dysfunction, immune dysregulation, microvascular thrombosis, mitochondrial injury, and maladaptive inflammatory signaling. Genetic susceptibility factors—including polymorphisms in ACE2, TMPRSS2, interferon signaling pathways, and APOL1 variants—likely influence both susceptibility to acute kidney injury during infection and subsequent long-term renal impairment.
Of particular clinical importance is the intersection between long COVID and metabolic disease. SARS-CoV-2 infection has been associated with increased incidence of new-onset diabetes mellitus, worsening glycemic control in preexisting diabetes, and acceleration of diabetic nephropathy. Disruption of pancreatic β-cell function, inflammatory insulin resistance, and dysregulation of renin–angiotensin–aldosterone signaling contribute to this metabolic phenotype.
This review synthesizes current evidence concerning the etiology, pathology, genomics, physiology, and clinical management of long COVID–associated kidney disease. Particular emphasis is placed on glomerular injury mechanisms, alterations in renal microcirculation, and therapeutic approaches to mitigate progression to chronic kidney disease. Understanding the renal consequences of persistent SARS-CoV-2 infection will be critical to the long-term management of millions of affected patients worldwide.
Introduction
In the aftermath of the global pandemic caused by SARS-CoV-2, a substantial subset of individuals experience persistent or newly emerging symptoms following recovery from the acute infection. This constellation of chronic manifestations—termed post-acute sequelae of SARS-CoV-2 infection (PASC) or long COVID—represents one of the most significant emerging challenges in contemporary medicine.
Long COVID is characterized by remarkable heterogeneity in clinical expression. Patients frequently report fatigue, dyspnea, cognitive impairment, dysautonomia, and metabolic disturbances months after initial infection. However, the kidney—an organ with extensive vascularization and high metabolic demand—has increasingly been recognized as particularly vulnerable to both direct viral injury and secondary inflammatory cascades.
Early in the pandemic, acute kidney injury (AKI) was observed in up to 30–40% of hospitalized patients with severe COVID-19. Subsequent longitudinal studies have demonstrated that even individuals with mild or moderate disease may develop persistent renal abnormalities months later, including reduced estimated glomerular filtration rate (eGFR), albuminuria, and progressive nephron loss.
These findings raise several fundamental questions regarding the nature of renal injury in long COVID:
- Is persistent kidney dysfunction the consequence of unresolved acute injury?
- Does SARS-CoV-2 establish chronic viral reservoirs within renal tissue?
- What genomic and molecular mechanisms determine susceptibility to renal complications?
- How does long COVID intersect with metabolic disorders such as diabetes mellitus?
Addressing these questions requires an integrated understanding of renal physiology, viral pathobiology, immune regulation, and metabolic homeostasis.
Epidemiology of Renal Sequelae in Long COVID
Population-based cohort studies conducted in the United States, Europe, and Asia have consistently demonstrated increased risk of renal impairment following SARS-CoV-2 infection.
Large analyses of electronic health records involving millions of individuals have identified several key observations:
- Increased incidence of chronic kidney disease among survivors of COVID-19
- Accelerated decline in glomerular filtration rate
- Increased frequency of albuminuria and proteinuria
- Elevated risk of end-stage kidney disease requiring dialysis
Notably, these outcomes are not restricted to patients who experienced severe acute illness. Even individuals managed entirely in the outpatient setting exhibit measurable reductions in renal function months after infection.
The magnitude of risk correlates with several factors:
- severity of initial infection
- presence of acute kidney injury during hospitalization
- preexisting metabolic disease
- advanced age
- genetic susceptibility
Older adults and individuals with hypertension, diabetes, or obesity appear particularly vulnerable.
Viral Tropism and Renal Susceptibility
The kidney possesses several biological features that make it particularly susceptible to viral infection and inflammatory injury.
SARS-CoV-2 enters host cells through interaction between the viral spike protein and the angiotensin-converting enzyme 2 (ACE2) receptor. This receptor is highly expressed in several renal cell populations, including:
- proximal tubular epithelial cells
- podocytes
- endothelial cells of glomerular capillaries
Co-expression of ACE2 with transmembrane protease serine 2 (TMPRSS2) facilitates viral entry and replication.
Several autopsy studies have demonstrated viral RNA and protein within renal tissue during acute infection. Viral particles have been detected in podocytes and proximal tubules using electron microscopy, though the interpretation of these findings remains debated.
Whether persistent viral reservoirs exist within renal tissue during long COVID remains uncertain. However, several lines of evidence suggest that viral persistence may contribute to chronic immune activation and endothelial dysfunction.
Genomic Susceptibility to Renal Complications
Host genetic variation appears to play an important role in determining susceptibility to severe COVID-19 and its long-term complications.
Multiple genomic loci have been implicated in COVID-19 severity through genome-wide association studies. Several of these loci have direct relevance to renal biology.
ACE2 Polymorphisms
Variation in the ACE2 gene may influence receptor expression and viral binding affinity. Individuals with higher ACE2 expression in renal tissue could theoretically experience increased viral entry and subsequent tissue injury.
APOL1 Risk Variants
Variants in the APOL1 gene—particularly common among individuals of African ancestry—have been strongly associated with certain forms of kidney disease, including focal segmental glomerulosclerosis and collapsing glomerulopathy.
During the pandemic, numerous cases of collapsing glomerulopathy associated with COVID-19 were reported, particularly among individuals carrying APOL1 risk alleles.
This entity has been termed:
COVID-19–associated nephropathy (COVAN)
The pathogenesis likely involves interferon-mediated activation of APOL1 expression, leading to podocyte injury and glomerular collapse.
Interferon Signaling Pathways
Genetic variants affecting interferon signaling influence the host antiviral response. Dysregulated interferon activity may contribute to persistent inflammation and endothelial injury in long COVID.
Renal Physiology and the Vulnerability of the Glomerulus
The glomerulus represents the fundamental filtration unit of the kidney. Each human kidney contains approximately one million nephrons, each composed of a glomerulus connected to a tubular system responsible for reabsorption and secretion.
The glomerular filtration barrier consists of three primary layers:
- Fenestrated endothelial cells
- Glomerular basement membrane
- Podocyte slit diaphragm
Together these structures maintain selective filtration, allowing water and small solutes to pass while retaining larger proteins such as albumin.
The glomerulus is exquisitely sensitive to disruptions in microvascular integrity, inflammatory signaling, and metabolic stress.
SARS-CoV-2 infection may compromise glomerular function through several mechanisms:
- endothelial inflammation
- complement activation
- microvascular thrombosis
- podocyte injury
- cytokine-mediated permeability changes
Even modest injury to these structures can result in albuminuria and progressive nephron loss.
Endothelial Dysfunction and Microvascular Injury
A hallmark feature of COVID-19 is widespread endothelial dysfunction. The vascular endothelium regulates blood flow, coagulation, immune signaling, and tissue oxygenation.
SARS-CoV-2 infection triggers a cascade of inflammatory events that disrupt endothelial homeostasis.
These processes include:
- activation of complement pathways
- release of proinflammatory cytokines
- platelet aggregation
- microthrombus formation
Within the kidney, these processes can impair glomerular perfusion and lead to ischemic injury.
Renal microvascular injury may persist long after viral clearance, contributing to chronic reductions in glomerular filtration rate.
Immune Dysregulation in Long COVID
Immune dysregulation is increasingly recognized as a central driver of long COVID pathophysiology.
Several immune abnormalities have been documented in affected individuals:
- persistent elevation of inflammatory cytokines
- altered T-cell activation patterns
- autoantibody production
- complement pathway activation
Chronic immune activation may sustain renal inflammation, promoting progressive fibrosis and nephron loss.
Additionally, autoantibodies targeting endothelial or renal antigens could contribute to ongoing tissue damage.
Mitochondrial Injury and Metabolic Dysfunction
Renal tubular cells possess exceptionally high metabolic demands due to their role in electrolyte transport and solute reabsorption.
Mitochondrial dysfunction has been observed in several studies of COVID-19 pathology. Viral infection can disrupt mitochondrial signaling, impair oxidative phosphorylation, and increase production of reactive oxygen species.
These processes may contribute to tubular injury and reduced renal energy metabolism, ultimately impairing filtration and reabsorption functions.
Transition to Chronic Kidney Disease
Persistent renal injury following COVID-19 may culminate in progressive chronic kidney disease.
Several mechanisms contribute to this transition:
- loss of functional nephrons
- compensatory hyperfiltration in remaining nephrons
- glomerular hypertension
- progressive fibrosis
Over time, these processes lead to declining eGFR and increasing risk of end-stage renal disease.
Renal Histopathology in Long COVID: Glomerular Injury, Microvascular Damage, and Tubular Pathology
Overview
Renal pathology associated with COVID-19 encompasses a spectrum of structural abnormalities involving the glomerulus, renal tubules, interstitium, and microvasculature. While acute kidney injury during the initial infection has been extensively documented, increasing evidence indicates that persistent renal injury contributes to the pathophysiology of Long COVID or post-acute sequelae of SARS-CoV-2 infection (PASC).
Autopsy studies and renal biopsy series have revealed several recurring histopathologic patterns, including:
- acute tubular injury
- collapsing glomerulopathy
- podocyte injury
- thrombotic microangiopathy
- endothelial inflammation
- interstitial fibrosis
These pathologic findings likely represent overlapping mechanisms driven by viral infection, immune dysregulation, complement activation, and microvascular injury.¹⁵–¹⁸
Acute Tubular Injury
The most common renal histologic abnormality observed in patients with COVID-19 is acute tubular injury (ATI).
The renal proximal tubule is particularly vulnerable to injury because of its high metabolic activity and dependence on mitochondrial oxidative phosphorylation. Viral infection, systemic inflammation, and hypoxia can disrupt tubular energy metabolism, leading to epithelial cell injury and necrosis.
Histologic features of ATI observed in COVID-19 include:
- loss of brush border in proximal tubular epithelial cells
- vacuolar degeneration
- tubular dilation
- epithelial cell detachment
- intraluminal debris
Electron microscopy studies have demonstrated mitochondrial swelling and fragmentation within tubular epithelial cells, suggesting severe metabolic stress.¹⁹
In patients who survive acute infection, incomplete recovery from tubular injury may contribute to progressive nephron loss and chronic kidney disease.
Podocyte Injury and Collapsing Glomerulopathy
Among the most striking glomerular pathologies associated with SARS-CoV-2 infection is collapsing glomerulopathy, a severe form of kidney injury characterized by collapse of the glomerular capillary tuft and proliferation of podocytes.
This lesion is strongly associated with variants in the APOL1 gene and has been described in multiple patients infected with SARS-CoV-2.²⁰
The condition has been termed:
COVID-19–Associated Nephropathy (COVAN)
Histologic characteristics include:
- segmental or global collapse of glomerular capillary loops
- hypertrophy and hyperplasia of podocytes
- protein reabsorption droplets
- microcystic tubular dilation
Electron microscopy often demonstrates widespread podocyte foot-process effacement.
Podocytes play a critical role in maintaining the integrity of the glomerular filtration barrier. Injury to these cells disrupts slit diaphragms and allows plasma proteins to leak into the urine, resulting in severe proteinuria.
Patients with collapsing glomerulopathy frequently develop rapid decline in renal function and may progress to dialysis-dependent kidney failure.
Endothelial Injury and Thrombotic Microangiopathy
The renal microvasculature is highly susceptible to endothelial injury during SARS-CoV-2 infection.
Autopsy studies have demonstrated widespread endothelial inflammation—termed endotheliitis—within renal capillaries and arterioles.²¹
These vascular abnormalities appear to be driven by several mechanisms:
- viral infection of endothelial cells
- activation of the Complement System
- cytokine-mediated inflammation
- platelet activation and microthrombus formation
In some patients, these processes culminate in thrombotic microangiopathy (TMA).
Histologic features of TMA include:
- endothelial swelling
- narrowing of capillary lumens
- fibrin thrombi within glomerular capillaries
- mesangiolysis
Microvascular occlusion impairs glomerular perfusion and can lead to ischemic injury of the filtration apparatus.
Persistent endothelial dysfunction has been proposed as a major contributor to renal impairment observed months after infection.
Complement Activation
Complement-mediated injury appears to play a central role in the pathogenesis of renal damage in COVID-19.
Multiple studies have identified deposition of complement components within renal tissue, including:
- C3
- C5b-9 (membrane attack complex)
Complement activation can injure endothelial cells and promote microvascular thrombosis.
The interaction between complement pathways and coagulation cascades may explain the frequent coexistence of inflammatory and thrombotic lesions in COVID-19 renal pathology.²²
Viral Presence in Renal Tissue
Several autopsy studies have identified RNA from Severe Acute Respiratory Syndrome Coronavirus 2 within kidney tissue using polymerase chain reaction and in situ hybridization.
In some cases, viral particles have been visualized within:
- podocytes
- proximal tubular epithelial cells
However, the interpretation of electron microscopy findings remains controversial. Some investigators argue that structures initially interpreted as viral particles may represent normal intracellular organelles.
Nevertheless, the presence of viral RNA within renal tissue suggests that direct viral infection may contribute to tissue injury during acute disease.
Whether persistent viral reservoirs exist in long COVID remains an area of active research.
Interstitial Inflammation and Fibrosis
Chronic kidney disease ultimately results from progressive interstitial fibrosis and nephron loss.
Histologic examination of renal tissue from patients recovering from severe COVID-19 has revealed varying degrees of:
- interstitial inflammatory infiltrates
- tubular atrophy
- collagen deposition
Fibrosis represents the final common pathway of many forms of kidney injury. Persistent inflammation following SARS-CoV-2 infection may accelerate fibrotic remodeling of renal tissue.
Over time, these structural changes reduce the number of functioning nephrons and lead to progressive decline in glomerular filtration rate.
Ultrastructural Findings
Electron microscopy has provided important insights into the cellular mechanisms of renal injury in COVID-19.
Common ultrastructural findings include:
- podocyte foot process effacement
- endothelial cell swelling
- mitochondrial disruption
- basement membrane irregularities
Mitochondrial injury appears particularly prominent in tubular epithelial cells, suggesting that metabolic dysfunction plays a significant role in disease pathogenesis.
Disruption of mitochondrial energy production may impair tubular reabsorption of sodium and other solutes, contributing to electrolyte abnormalities frequently observed in COVID-19 patients.
Relationship Between Acute Kidney Injury and Long-Term Renal Dysfunction
A large body of evidence indicates that acute kidney injury during the initial infection strongly predicts subsequent development of chronic kidney disease.
However, emerging data suggest that even individuals who did not experience clinically recognized AKI may develop progressive renal dysfunction.
Several mechanisms may explain this phenomenon:
- subclinical nephron injury
- persistent endothelial dysfunction
- chronic immune activation
- microvascular remodeling
These processes may slowly impair renal function over months or years following infection.
Implications for Long-COVID Clinical Monitoring
Recognition of these histopathologic mechanisms underscores the importance of long-term renal monitoring in patients recovering from COVID-19.
Recommended surveillance measures include:
- periodic assessment of estimated glomerular filtration rate
- measurement of urine albumin-to-creatinine ratio
- blood pressure monitoring
- screening for metabolic abnormalities
Early detection of renal injury may permit initiation of therapies capable of slowing progression to chronic kidney disease.
Metabolic Dysregulation in Long COVID: Pancreatic Injury, Diabetes, and Renal Consequences
Introduction
Metabolic dysfunction has emerged as a major component of Long COVID, with increasing evidence demonstrating that infection with Severe Acute Respiratory Syndrome Coronavirus 2 may precipitate new-onset diabetes or exacerbate preexisting metabolic disease. These metabolic abnormalities have profound implications for renal physiology and may accelerate the progression of chronic kidney disease.
Observational studies conducted during the pandemic have demonstrated that survivors of COVID-19 exhibit significantly increased risks of developing Type 2 Diabetes within months after infection.²³ In some cohorts, the incidence of new diabetes diagnoses among COVID-19 survivors has been estimated to be 30–40% higher than in matched control populations without prior infection.
The mechanisms underlying this phenomenon appear multifactorial, involving direct viral injury to pancreatic tissue, inflammatory insulin resistance, alterations in adipose metabolism, and dysregulation of the Renin–Angiotensin–Aldosterone System (RAAS).
Because diabetes is among the most important drivers of kidney disease worldwide, the emergence of COVID-associated metabolic dysfunction carries substantial long-term implications for renal health.
Viral Infection of Pancreatic Tissue
Several experimental and histopathologic studies have demonstrated that pancreatic endocrine cells express the ACE2 receptor required for viral entry.²⁴
ACE2 expression has been identified in:
- pancreatic β-cells
- pancreatic ductal epithelium
- microvascular endothelial cells
This distribution suggests that pancreatic tissue may be directly susceptible to SARS-CoV-2 infection.
In vitro studies using human pancreatic organoids have demonstrated that SARS-CoV-2 can infect insulin-producing β-cells, leading to impaired insulin secretion and cellular apoptosis.²⁵
Autopsy analyses of patients who died from COVID-19 have detected viral RNA and protein within pancreatic tissue, further supporting the possibility of direct pancreatic infection.
Damage to β-cells reduces insulin production and may contribute to hyperglycemia and diabetes.
Inflammatory Insulin Resistance
Beyond direct viral injury, systemic inflammation plays a central role in metabolic dysregulation following COVID-19.
Acute infection triggers a robust immune response characterized by elevated levels of proinflammatory cytokines such as:
- interleukin-6
- tumor necrosis factor α
- interleukin-1β
These cytokines interfere with insulin signaling pathways in skeletal muscle, adipose tissue, and liver.
Persistent low-grade inflammation observed in long COVID may therefore sustain insulin resistance long after viral clearance.²⁶
Insulin resistance leads to increased hepatic glucose production and reduced peripheral glucose uptake, resulting in chronic hyperglycemia.
Dysregulation of the Renin–Angiotensin System
SARS-CoV-2 interacts directly with the ACE2 receptor, a key regulator of the Renin–Angiotensin–Aldosterone System.
Under physiologic conditions, ACE2 converts angiotensin II into angiotensin-(1–7), a peptide with vasodilatory and anti-inflammatory properties.
Viral binding to ACE2 leads to downregulation of receptor expression, resulting in increased levels of angiotensin II.
Elevated angiotensin II promotes:
- vasoconstriction
- oxidative stress
- inflammation
- fibrosis
Within the kidney, these effects may increase intraglomerular pressure and accelerate structural damage to the filtration barrier.
Hyperglycemia and Glomerular Injury
Persistent hyperglycemia is a major driver of renal injury. In the context of long COVID, metabolic dysregulation may exacerbate preexisting kidney disease or initiate new forms of glomerular pathology.
Several mechanisms link hyperglycemia to glomerular damage:
Advanced Glycation End Products
Elevated glucose concentrations promote the formation of advanced glycation end products (AGEs). These molecules accumulate within the Glomerular Basement Membrane, altering its structural integrity and permeability.
Oxidative Stress
Hyperglycemia increases production of reactive oxygen species within renal cells, damaging podocytes and endothelial cells.
Hemodynamic Changes
Diabetes alters glomerular hemodynamics by increasing afferent arteriolar dilation. This process raises intraglomerular pressure, leading to hyperfiltration and mechanical stress on the filtration barrier.
Over time, these processes contribute to progressive nephron loss.
Diabetic Nephropathy in Long COVID
The intersection between COVID-associated metabolic dysregulation and renal injury may accelerate the development of Diabetic Nephropathy.
Diabetic nephropathy is characterized by several structural abnormalities:
- mesangial expansion
- thickening of the glomerular basement membrane
- podocyte loss
- glomerulosclerosis
Persistent hyperglycemia also promotes inflammation and fibrosis within renal tissue.
Patients with long COVID who develop diabetes may therefore experience accelerated progression to chronic kidney disease.
Interaction with Preexisting Diabetes
Individuals with established Type 2 Diabetes appear particularly vulnerable to the renal consequences of COVID-19.
Several factors contribute to this increased risk:
- baseline endothelial dysfunction
- chronic inflammation
- preexisting glomerular hyperfiltration
- impaired immune regulation
COVID-19 may exacerbate these processes, leading to more rapid deterioration of renal function.
Large cohort studies have demonstrated that diabetic patients infected with SARS-CoV-2 exhibit higher rates of:
- acute kidney injury
- albuminuria
- long-term decline in eGFR
Therapeutic Management
Given the complex interactions between long COVID, metabolic disease, and renal dysfunction, therapeutic management requires an integrated approach.
Glycemic Control
Strict glycemic control remains a cornerstone of preventing diabetic kidney disease. Recommended strategies include:
- lifestyle modification
- insulin therapy when necessary
- oral antihyperglycemic agents
Several classes of medications may confer additional renal protection.
SGLT2 Inhibitors
Drugs in the class of SGLT2 inhibitors reduce glucose reabsorption in the proximal tubule, thereby lowering blood glucose levels and improving renal outcomes.
These agents also reduce intraglomerular pressure by restoring tubuloglomerular feedback, which may slow progression of chronic kidney disease.
GLP-1 Receptor Agonists
Another class of medications with potential benefit includes GLP-1 receptor agonists. These drugs improve glycemic control and may exert anti-inflammatory effects.
RAAS Blockade
Inhibition of the Renin–Angiotensin–Aldosterone System using ACE inhibitors or angiotensin receptor blockers remains a cornerstone therapy for diabetic nephropathy.
These medications reduce intraglomerular pressure and limit proteinuria.
Implications for Long-Term Renal Outcomes
The convergence of SARS-CoV-2 infection, metabolic dysregulation, and renal injury raises concern regarding a potential future increase in chronic kidney disease prevalence.
Given the large global population infected during the pandemic, even modest increases in diabetes and kidney disease incidence could translate into millions of new cases of renal impairment.
Long-term surveillance and early intervention will therefore be essential components of post-pandemic healthcare.
Renal Physiology and Glomerular Hemodynamics in Long COVID
Normal Physiology of Glomerular Filtration
The kidney performs essential functions in the regulation of fluid balance, electrolyte homeostasis, and metabolic waste excretion. Central to these processes is the Glomerular Filtration mechanism, in which plasma is filtered across a specialized capillary network within the renal corpuscle.
Each kidney contains approximately one million nephrons, the functional units responsible for filtration and tubular processing of plasma. The glomerulus consists of a tuft of capillaries lined by fenestrated endothelial cells, supported by mesangial cells, and enveloped by visceral epithelial cells known as Podocytes.
The filtration barrier is composed of three highly specialized structures:
- fenestrated glomerular endothelium
- the Glomerular Basement Membrane
- the slit diaphragm formed between podocyte foot processes
These structures work together to regulate the selective passage of water and solutes while preventing the loss of large plasma proteins such as albumin.
Glomerular filtration rate (GFR) is determined primarily by the balance of hydrostatic and oncotic pressures across the glomerular capillary wall. Regulation of afferent and efferent arteriolar tone plays a critical role in maintaining stable filtration despite fluctuations in systemic blood pressure.
Alterations in Glomerular Hemodynamics After SARS-CoV-2 Infection
Infection with Severe Acute Respiratory Syndrome Coronavirus 2 may disrupt the delicate balance of glomerular hemodynamics through several mechanisms.
One major pathway involves dysregulation of the Renin–Angiotensin–Aldosterone System (RAAS). Viral binding to the ACE2 receptor leads to downregulation of ACE2 activity, thereby altering the balance between angiotensin II and angiotensin-(1–7).
Elevated angiotensin II levels promote efferent arteriolar constriction and increased intraglomerular pressure. While this mechanism may initially preserve filtration in the setting of systemic illness, sustained elevation of intraglomerular pressure can cause structural injury to the filtration barrier.
Chronic glomerular hypertension is a well-established driver of progressive kidney disease.
Endothelial Dysfunction and Impaired Microvascular Regulation
Another critical determinant of renal physiology is the integrity of the microvascular endothelium. Endothelial cells regulate vascular tone through the release of vasoactive mediators such as nitric oxide and prostacyclin.
COVID-19 has been widely recognized as a disease characterized by widespread endothelial injury. Viral infection and inflammatory cytokines impair endothelial nitric oxide production and promote a prothrombotic state.²⁷
Within the kidney, endothelial dysfunction may produce several hemodynamic consequences:
- impaired autoregulation of renal blood flow
- increased vascular resistance
- microvascular thrombosis
These alterations reduce oxygen delivery to renal tissue and may contribute to chronic ischemic injury.
Tubuloglomerular Feedback
A key regulatory mechanism controlling glomerular filtration is Tubuloglomerular Feedback, in which specialized cells within the macula densa monitor sodium chloride concentration in the distal tubule.
When sodium delivery to the macula densa increases, afferent arteriolar constriction reduces glomerular filtration rate. Conversely, reduced sodium delivery promotes vasodilation and increased filtration.
SARS-CoV-2–related tubular injury may disrupt this feedback mechanism. Damage to proximal tubular epithelial cells alters sodium reabsorption, thereby affecting signals transmitted to the macula densa.
Disruption of tubuloglomerular feedback can lead to unstable glomerular hemodynamics and contribute to progressive nephron injury.
Autonomic Nervous System Dysregulation
Long COVID has been associated with significant abnormalities in autonomic nervous system function, including orthostatic intolerance and Postural Orthostatic Tachycardia Syndrome.
The kidneys receive extensive sympathetic innervation, which regulates renin release, renal vascular resistance, and sodium reabsorption.
Persistent sympathetic activation may therefore influence renal physiology through several mechanisms:
- increased renin secretion
- enhanced sodium retention
- elevated systemic blood pressure
These changes may contribute to the development of hypertension and exacerbate renal injury.
Neuroendocrine Regulation of Renal Function
In addition to autonomic pathways, renal physiology is influenced by several hormonal systems.
These include:
- the Renin–Angiotensin–Aldosterone System
- antidiuretic hormone (vasopressin)
- natriuretic peptides
- the sympathetic nervous system
Evidence suggests that SARS-CoV-2 infection may disrupt neuroendocrine regulation through inflammatory injury to hypothalamic and brainstem structures.
Alterations in vasopressin signaling have been proposed as a contributor to electrolyte disturbances frequently observed in COVID-19 patients, including hyponatremia.
Mitochondrial Dysfunction and Renal Energy Metabolism
Renal tubular cells have exceptionally high metabolic demands, particularly within the proximal tubule where active transport of sodium, glucose, and other solutes occurs.
Emerging evidence suggests that mitochondrial dysfunction plays an important role in long-COVID pathophysiology.
Studies have demonstrated:
- mitochondrial fragmentation
- impaired oxidative phosphorylation
- increased production of reactive oxygen species
These abnormalities reduce ATP availability and impair tubular transport processes.
Chronic mitochondrial dysfunction may therefore contribute to persistent renal impairment even after resolution of acute infection.
Persistent Inflammation and Fibrotic Remodeling
Another important mechanism underlying chronic kidney disease is fibrotic remodeling of renal tissue.
Persistent immune activation in long COVID may stimulate fibroblast proliferation and deposition of extracellular matrix proteins within the renal interstitium.
Several profibrotic mediators have been implicated, including:
- transforming growth factor β
- connective tissue growth factor
- angiotensin II
These pathways promote progressive scarring and loss of functional nephrons.
Biomarkers of Renal Injury in Long COVID
Identification of sensitive biomarkers may facilitate early detection of renal injury in patients recovering from COVID-19.
Potential biomarkers include:
- cystatin C
- neutrophil gelatinase–associated lipocalin
- kidney injury molecule-1
These markers may detect renal injury before significant changes in glomerular filtration rate become apparent.
Early detection may allow timely initiation of renoprotective therapies.
Clinical Management of Long-COVID Renal Disease
Early Detection and Monitoring
Given the heterogeneous renal manifestations of long COVID, early recognition and surveillance are critical. Recommendations for post-COVID patients include:
- Laboratory evaluation: serum creatinine, cystatin C, estimated glomerular filtration rate (eGFR), and urine albumin-to-creatinine ratio.³¹
- Blood pressure monitoring: hypertension is both a risk factor and consequence of renal injury.³²
- Metabolic assessment: fasting glucose, HbA1c, and lipid profiles to detect new-onset diabetes or metabolic dysregulation.²³
Early detection of subclinical renal injury allows timely initiation of nephroprotective therapies, potentially preventing progression to chronic kidney disease.
Pharmacologic Management
Management strategies are designed to mitigate ongoing injury and preserve renal function.
RAAS Inhibition
ACE inhibitors and angiotensin receptor blockers remain foundational therapies. By reducing intraglomerular pressure and proteinuria, RAAS blockade slows the progression of both COVID-associated kidney disease and diabetic nephropathy.³³
SGLT2 Inhibitors
Sodium-glucose co-transporter 2 (SGLT2) inhibitors provide dual benefits:
- glycemic control
- renoprotection via reduction of intraglomerular hypertension and preservation of tubular energy homeostasis.³⁴
Emerging studies suggest that SGLT2 inhibitors may also modulate inflammatory pathways implicated in long COVID.
GLP-1 Receptor Agonists
Glucagon-like peptide-1 (GLP-1) receptor agonists improve glycemic control, reduce weight, and may exert anti-inflammatory and anti-fibrotic effects on renal tissue.³⁵
Anti-inflammatory and Anti-complement Therapy
In selected patients, persistent inflammation or complement-mediated injury may warrant targeted therapy:
- corticosteroids for severe inflammatory flares
- complement inhibitors in thrombotic microangiopathy (TMA) associated with renal injury.³⁶
These interventions remain under investigation and should be individualized based on risk-benefit assessment.
Renal Replacement Therapy and Dialysis
For patients progressing to severe kidney failure, renal replacement therapy (RRT) may be required. Key considerations include:
- Hemodialysis: preferred for acute and chronic settings, particularly when rapid solute removal is required.³⁷
- Peritoneal dialysis: may be advantageous in resource-limited settings or for home-based therapy.³⁸
- Timing: early initiation in rapidly progressive AKI can prevent further metabolic derangements.
Evidence suggests that COVID-19 patients undergoing RRT may exhibit higher morbidity and mortality, underscoring the importance of preventive measures and early intervention.
Lifestyle and Supportive Measures
Non-pharmacologic interventions remain critical in mitigating renal injury:
- Dietary management: low-sodium diet, protein moderation, and glycemic control.
- Hydration: adequate fluid intake to maintain renal perfusion.
- Physical activity: tailored exercise to improve cardiovascular and metabolic health.
- Vaccination and infection prevention: to reduce risk of subsequent viral insults that could exacerbate renal injury.³⁹
Emerging Therapies and Experimental Interventions
Long COVID has spurred investigation into novel therapies targeting the underlying pathophysiology of renal injury.
Anti-fibrotic Agents
Drugs targeting transforming growth factor β (TGF-β) and connective tissue growth factor (CTGF) are under investigation for their ability to reduce renal fibrosis.⁴⁰
Mitochondrial Modulators
Given the role of mitochondrial dysfunction in tubular injury, therapies aimed at restoring mitochondrial bioenergetics, such as nicotinamide adenine dinucleotide (NAD⁺) precursors, may provide benefit.⁴¹
Immunomodulatory Therapy
For persistent immune activation and complement-mediated injury, targeted immunotherapy—including monoclonal antibodies against complement components—represents an area of active research.⁴²
Long-Term Outcomes and Epidemiologic Implications
The interplay between long COVID, diabetes, and renal injury has significant implications for public health:
- Survivors of COVID-19 may experience accelerated onset of chronic kidney disease, particularly in those with preexisting risk factors.²
- Population-level modeling predicts increased prevalence of albuminuria, proteinuria, and reduced GFR in post-COVID cohorts.⁴³
- Early identification, risk stratification, and nephroprotective interventions are essential to prevent a surge in end-stage renal disease.
Longitudinal studies are needed to quantify the burden of chronic kidney disease attributable to SARS-CoV-2 infection and to guide health policy and resource allocation.
Future Research Directions
Several knowledge gaps remain:
- Mechanisms of persistent viral or immunologic injury: Does SARS-CoV-2 establish renal reservoirs in long COVID?
- Biomarkers of early renal injury: Identification of sensitive and specific markers for subclinical nephron damage.
- Long-term effects of COVID-associated diabetes: Determining whether post-COVID diabetes follows the natural course of type 2 diabetes or represents a distinct phenotype.
- Efficacy of emerging therapies: Clinical trials targeting mitochondrial function, fibrosis, and complement pathways.
- Impact of vaccination and antiviral therapy: Evaluating whether early viral suppression mitigates long-term renal complications.
Addressing these questions is critical to improve outcomes for the growing population of long-COVID survivors.
Discussion
The current body of evidence demonstrates that Severe Acute Respiratory Syndrome Coronavirus 2 infection exerts profound and sustained effects on renal physiology, particularly within the context of Long COVID. The kidney emerges as both a target organ for direct viral injury and a site of secondary pathology resulting from systemic inflammation, endothelial dysfunction, and metabolic dysregulation.¹⁵,²³,²⁷
Autopsy and biopsy studies reveal a spectrum of renal injury patterns, ranging from acute tubular injury to podocyte injury and collapsing glomerulopathy, often accompanied by microvascular thrombosis and interstitial fibrosis.¹⁶–¹⁸,²⁰,²¹ These structural abnormalities reflect the interplay between direct viral effects, host genetic susceptibility (e.g., ACE2 and APOL1 polymorphisms), and dysregulated immune and complement pathways.⁴–⁶,²²
Long-term renal dysfunction in survivors of COVID-19 is further amplified by the virus’s impact on glucose metabolism. Post-infectious hyperglycemia, new-onset diabetes, and exacerbation of preexisting diabetes accelerate podocyte injury, mesangial expansion, and glomerular basement membrane thickening, key features of diabetic nephropathy.²³–²⁶,³⁴,³⁵ The convergence of hyperglycemia, RAAS dysregulation, and microvascular endothelial dysfunction establishes a “perfect storm” for progressive nephron loss.
Disruptions in renal hemodynamics and neuroendocrine regulation further contribute to chronic injury. Dysregulated tubuloglomerular feedback, persistent sympathetic activation, and altered vasopressin and natriuretic peptide signaling compromise glomerular filtration stability, while mitochondrial dysfunction impairs tubular energy metabolism and sodium reabsorption.²⁷–³¹ Collectively, these mechanisms explain the observation that even patients without clinically apparent acute kidney injury during initial infection may develop long-term renal impairment.²
The clinical implications of these findings are substantial. The population-level burden of chronic kidney disease is likely to increase as a consequence of the pandemic, particularly among individuals with preexisting metabolic risk factors.³,⁴³ Early detection through laboratory monitoring, utilization of renoprotective pharmacologic agents (RAAS inhibitors, SGLT2 inhibitors, GLP-1 receptor agonists), and attention to lifestyle modification represent the foundation of long-COVID renal management.³¹–³⁵,³⁹
Emerging therapies targeting fibrosis, complement activation, and mitochondrial dysfunction hold promise but require rigorous clinical evaluation.⁴⁰–⁴² Furthermore, the role of vaccination, antiviral therapy, and early intervention in mitigating long-term renal sequelae warrants further investigation.
Conclusion
Renal injury represents a central component of long COVID, encompassing both direct viral cytopathy and secondary effects of systemic inflammation, endothelial dysfunction, and metabolic derangements. Glomerular injury—including podocyte loss, collapsing glomerulopathy, and microvascular thrombosis—intersects with dysregulated glucose metabolism to accelerate the development of chronic kidney disease, particularly in individuals with new-onset or preexisting diabetes.
Management of long-COVID renal disease requires an integrated approach, combining vigilant surveillance, glycemic control, RAAS inhibition, and emerging therapies aimed at modulating inflammation, complement activation, and fibrosis. Population-level strategies to monitor renal outcomes and mitigate risk are critical to prevent a post-pandemic surge in kidney disease.
Future research should focus on elucidating mechanisms of persistent viral or immune-mediated injury, identifying sensitive biomarkers for early detection, and testing novel interventions to preserve nephron function. Understanding the complex interplay between SARS-CoV-2 infection, metabolic dysfunction, and renal pathology is essential for optimizing outcomes in the millions of long-COVID survivors worldwide.
References
- Nalbandian A, Sehgal K, Gupta A, et al. Post-acute COVID-19 syndrome. Nat Med. 2021.
- Al-Aly Z, Xie Y, Bowe B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature. 2021.
- Bowe B, Xie Y, Al-Aly Z. Kidney outcomes in long COVID. J Am Soc Nephrol. 2021.
- Ellinghaus D, et al. Genomewide association study of severe COVID-19. N Engl J Med. 2020.
- Larsen CP, et al. Collapsing glomerulopathy in COVID-19. Kidney International Reports. 2020.
- Zhang Q, et al. Inborn errors of interferon immunity in COVID-19. Science. 2020.
- Rubino F, et al. New-onset diabetes in COVID-19. N Engl J Med. 2020.
- Gupta A, et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020.
- Davis HE, et al. Characterizing long COVID. EClinicalMedicine. 2021.
- Hirsch JS, et al. Acute kidney injury in hospitalized patients with COVID-19. Kidney International. 2020.
- Hamming I, et al. Tissue distribution of ACE2. J Pathol. 2004.
- Hoffmann M, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2. Cell. 2020.
- Puelles VG, et al. Multiorgan viral tropism of SARS-CoV-2. N Engl J Med. 2020.
- Varga Z, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020.
- Su H, Yang M, Wan C, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19. Kidney Int. 2020.
- Golmai P, Larsen CP, DeVita MV, et al. Histopathologic and ultrastructural findings in COVID-19 kidney biopsy specimens. J Am Soc Nephrol. 2020.
- Sharma P, Uppal NN, Wanchoo R, et al. COVID-19-associated kidney injury. Kidney Int Rep. 2020.
- Peleg Y, Kudose S, D’Agati V, et al. Acute kidney injury due to collapsing glomerulopathy following COVID-19 infection. Kidney Int Rep. 2020.
- Farkash EA, Wilson AM, Jentzen JM. Ultrastructural evidence for direct renal infection with SARS-CoV-2. Lancet. 2020.
- Larsen CP, Bourne TD, Wilson JD, et al. Collapsing glomerulopathy in a patient with COVID-19. Kidney Int Rep. 2020.
- Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020.
- Magro C, Mulvey JJ, Berlin D, et al. Complement activation in microvascular injury in COVID-19. Transl Res. 2020.
- Xie Y, Al-Aly Z. Risks and burdens of incident diabetes in long COVID. Lancet Diabetes Endocrinol. 2022.
- Kusmartseva I, Wu W, Syed F, et al. Expression of SARS-CoV-2 entry factors in the pancreas. Cell Metabolism. 2020.
- Müller JA, Groß R, Conzelmann C, et al. SARS-CoV-2 infects pancreatic β-cells. Nature Metabolism. 2021.
- Montefusco L, Ben Nasr M, D’Addio F, et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nature Metabolism. 2021.
- Libby P, Luscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020.
- Batlle D, Soler MJ, Sparks MA, et al. Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J Am Soc Nephrol. 2020.
- Ronco C, Reis T, Husain-Syed F. Management of acute kidney injury in patients with COVID-19. Lancet Respir Med. 2020.
- Nadim MK, Forni LG, Mehta RL, et al. COVID-19–associated acute kidney injury. Nat Rev Nephrol. 2020.
- Cheng Y, Luo R, Wang K, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020.
- Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020.
- Inker LA, et al. RAAS blockade in COVID-19 and renal outcomes. N Engl J Med. 2021.
- Heerspink HJL, et al. SGLT2 inhibitors in diabetic kidney disease. Lancet Diabetes Endocrinol. 2020.
- Gerstein HC, et al. GLP-1 receptor agonists and renal outcomes. N Engl J Med. 2019.
- Magro C, et al. Complement in COVID-19–associated microvascular injury. Transl Res. 2020.
- Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. 2019.
- Li PK, et al. Peritoneal dialysis in COVID-19. Kidney Int Rep. 2020.
- Izzedine H, et al. Management of chronic kidney disease post-COVID-19. Kidney Int. 2021.
- Liu Y, et al. Anti-fibrotic therapies in kidney disease. Nat Rev Nephrol. 2018.
- Hall AM, et al. Mitochondrial dysfunction in renal tubular cells. J Am Soc Nephrol. 2019.
- Noris M, Remuzzi G. Complement-targeted therapies in kidney disease. Nat Rev Nephrol. 2020.
- Xie Y, Bowe B, Al-Aly Z. Long-term kidney outcomes after COVID-19. Nat Med. 2022.
- Bowe B, Xie Y, Xu E, Al-Aly Z. Kidney outcomes in long COVID. J Am Soc Nephrol. 2021.
- Rossing K, et al. Diabetic nephropathy: epidemiology, mechanisms, and management. Lancet Diabetes Endocrinol. 2021.
- Cheng Y, Luo R, Wang K, et al. Kidney disease and mortality in COVID-19. Kidney Int. 2020.
- Siddiqi HK, Mehra MR. COVID-19 illness in native kidneys and transplant recipients. Lancet Respir Med. 2020.
- Xu E, Xie Y, Al-Aly Z. Post-acute sequelae of COVID-19 and long-term renal risk. Nat Med. 2022.