- Authors: Bruno Bordallo, Mozart Bellas, Arthur Fernandes Cortez, Matheus Vieira & Marcelo Pinheiro Advances in Rheumatology volume 60, Article number: 50 (2020) Cite this article
Abstract
The COVID-19 outbreak caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has become a global major concern. In this review, we addressed a theoretical model on immunopathogenesis associated with severe COVID-19, based on the current literature of SARS-CoV-2 and other epidemic pathogenic coronaviruses, such as SARS and MERS. Several studies have suggested that immune dysregulation and hyperinflammatory response induced by SARS-CoV-2 are more involved in disease severity than the virus itself.
Immune dysregulation due to COVID-19 is characterized by delayed and impaired interferon response, lymphocyte exhaustion and cytokine storm that ultimately lead to diffuse lung tissue damage and posterior thrombotic phenomena.
Considering there is a lack of clinical evidence provided by randomized clinical trials, the knowledge about SARS-CoV-2 disease pathogenesis and immune response is a cornerstone to develop rationale-based clinical therapeutic strategies. In this narrative review, the authors aimed to describe the immunopathogenesis of severe forms of COVID-19.
Background
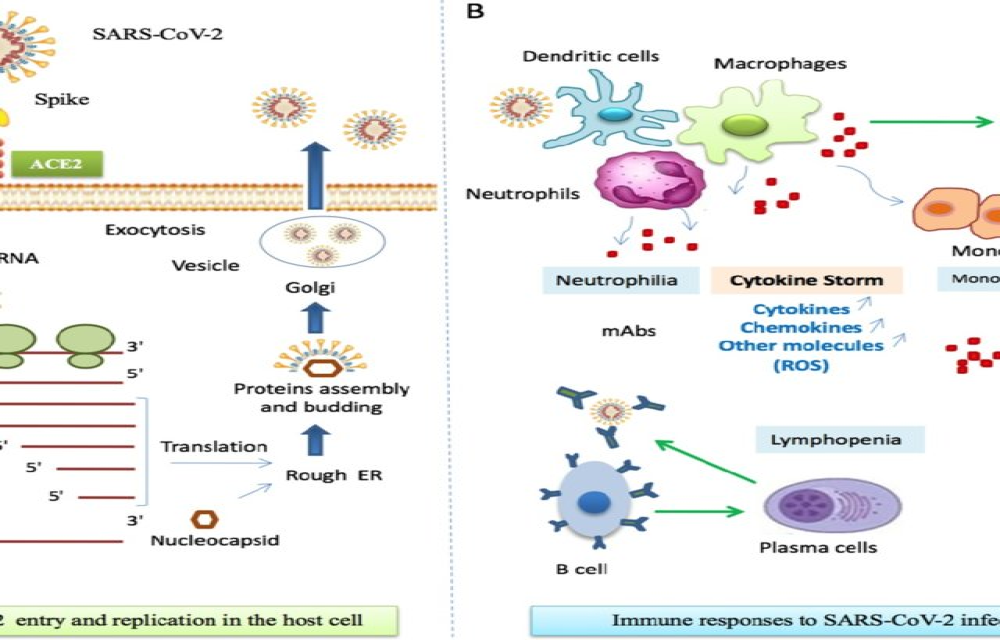
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a positive-sense single-stranded RNA-enveloped virus, is the causative agent of coronavirus disease 2019 (COVID-19), being first identified in Wuhan, China, in December 2019. Previously, other epidemic coronavirus such as severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 and the middle-east respiratory syndrome coronavirus (MERS-CoV) in 2012, had serious impact on human health and warned the world about the possible reemergence of new pathogenic strains [1]. Despite being a new virus, several common morpho-functional characteristics have been reported between SARS-CoV and the SARS-CoV-2, including the interaction of the viral spike (S) glycoprotein with the human angiotensin converting enzyme 2 (ACE2). These similarities may help understanding some pathophysiological mechanisms and pointing out possible therapeutic targets.
The first step for SARS-CoV-2 entry into the host cell is the interaction between the S glycoprotein and ACE2 on cell surface. Since the latter acts as a viral receptor, the virus will only infect ACE2 expressing cells, notably type II pneumocytes. These cells represent 83% of the ACE2-expressing cells in humans, but cells from other tissues and organs, such as heart, kidney, intestine and endothelium, can also express this receptor [2]. A host type 2 transmembrane serine protease, TMPRSS2, facilitates virus entry by priming S glycoprotein. TMPRSS2 entails S protein in subunits S1/S2 and S2´, allowing viral and cellular membrane fusion driven by S2 subunit [3]. Once inside the cell viral positive sense single strand RNA is translated into polyproteins that will form the replicase-transcriptase complex. This complex function as a viral factory producing new viral RNA and viral proteins for viral function and assembly [4]. Considering these particularities, the infection first begins on upper respiratory tract mucosa and then reaches the lungs. The primary tissue damage is related to the direct viral cytopathic effects. At this stage, the virus has the potential to evade the immune system, where an inadequate innate immune response can occur, depending on the viral load and other unknown genetic factors. Subsequently, tissue damage is induced by additional mechanisms derived from a dysregulated adaptive immune response [5].
Although most of COVID-19 cases have a mild clinical course, up to 14% can evolve to a severe form, with respiratory rate ≥ 30/min, hypoxemia with pulse oxygen saturation ≤ 93%, partial pressure of arterial oxygen to fraction of inspired oxygen ratio < 300 and/or pulmonary infiltrates involving more than 50% of lung parenchyma within 24 to 48 h. Up to 5% of the cases can be critical, evolving with respiratory failure, septic shock and/or multiple organ dysfunction, presumably driven by a cytokine storm [6]. Host characteristics, including aging (immunosenescence) and comorbidities (hypertension, diabetes mellitus, lung and heart diseases) may influence the course of the disease [7]. The false paradox between inflammation and immunodeficiency is highlighted by the severe form of COVID-19. Thus, severe pneumonia caused by SARS-CoV-2 is marked by immune system dysfunction and hyperinflammation leading to acute respiratory distress syndrome (ARDS), macrophage activation, hypercytokinemia and coagulopathy [8].
Herein, we aim to review the factors related to the dysregulated immune response against the SARS-CoV-2, along with its relation with severe forms of COVID-19, namely ARDS and cytokine storm (CS).
For More Information: https://advancesinrheumatology.biomedcentral.com/articles/10.1186/s42358-020-00151-7