Authors: Setu M. Vora,1,2Judy Lieberman,2,3 and Hao Wu1,2
Abstract
The COVID-19 pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), results in life-threatening disease in a minority of patients, especially elderly people and those with co-morbidities such as obesity and diabetes. Severe disease is characterized by dysregulated cytokine release, pneumonia and acute lung injury, which can rapidly progress to acute respiratory distress syndrome, disseminated intravascular coagulation, multisystem failure and death. However, a mechanistic understanding of COVID-19 progression remains unclear. Here we review evidence that SARS-CoV-2 directly or indirectly activates inflammasomes, which are large multiprotein assemblies that are broadly responsive to pathogen-associated and stress-associated cellular insults, leading to secretion of the pleiotropic IL-1 family cytokines (IL-1β and IL-18), and pyroptosis, an inflammatory form of cell death. We further discuss potential mechanisms of inflammasome activation and clinical efforts currently under way to suppress inflammation to prevent or ameliorate severe COVID-19.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for COVID-19, has so far infected more than 190 million people and caused death of more than 4.1 million people worldwide. The virus primarily infects the respiratory tract, causing fever, sore throat, anosmia and dyspnoea, but its tissue tropism still remains to be fully understood. As many as 10–15% of patients develop severe pneumonia, with some cases progressing to hypoxia and acute respiratory distress syndrome (ARDS), which requires mechanical ventilation in a critical care setting and has high mortality. Patients can also develop multi-organ failure, acute kidney injury and disseminated intravascular coagulation, among a host of other disorders1–11. Aside from supportive care, only a few treatments have been approved for COVID-19, and their reduction of mortality has been limited12–14. Although several vaccines against SARS-CoV-2 have been approved and are being administered internationally, there will still be a significant number of infections owing to people who are not vaccinated in regions with inadequate access or acceptance of vaccination. In addition, while global vaccination efforts strive to meet the challenge of ending the pandemic, the appearance of immune-evasive viral variants and the unlikelihood of reaching immediate herd immunity underscore the continued need for additional treatments mitigating disease progression15–19.
Most researchers agree that an inappropriate hyperinflammatory response lies at the root of many severe cases of COVID-19, driven by overexuberant inflammatory cytokine release. Consistently, co-morbidities, such as obesity, diabetes, heart disease, hypertension and ageing, which are prognostic of poor outcome, are associated with high basal inflammation7,11,20,21. It has been proposed since the beginning of the pandemic that these co-morbidities and the ensuing hyperinflammatory response may be aetiologically linked through overactive inflammasome signaling, which may account for the association of these co-morbidities with severe COVID-19 in the context of chronic inflammation as well as for COVID-19 progression in the context of a robust acute inflammatory response to infection22–29. However, many of the studies that seek to understand the immune response to SARS-CoV-2 are based on RNA sequencing, often of thawed cells, and infected, activated or dying cells do not survive freeze–thaw well, which could skew results. Moreover, inflammasome activation does not directly induce transcriptional responses, and its detection is less straightforward than that of most other signaling pathways. Nonetheless, several studies are now accumulating that support direct (infection-induced) and indirect inflammasome activation and the critical role of inflammasomes in severe COVID-19. Here we discuss the available evidence, potential mechanisms and the implications for therapy.
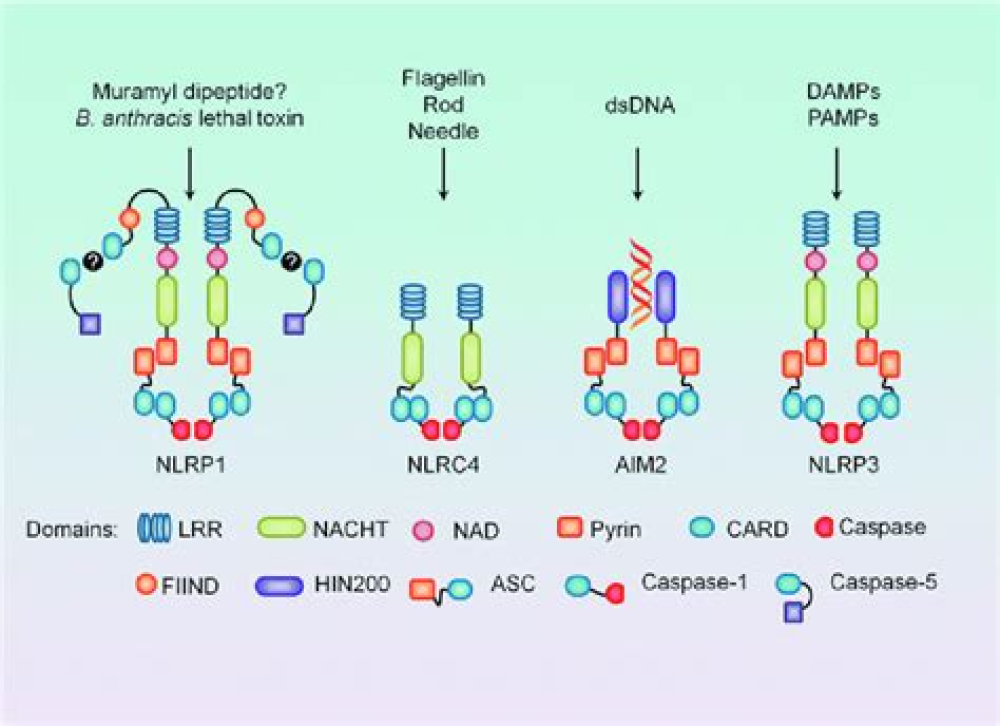
Key to inflammation and innate immunity, are large, micrometer-scale multiprotein cytosolic complexes that assemble in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) and trigger proinflammatory cytokine release as well as pyroptosis, a proinflammatory lytic cell death30,31 (Fig. 1). Upon activation by PAMPs or DAMPs, canonical inflammasome sensors — mainly in monocytes, macrophages and barrier epithelial cells — oligomerize and recruit the adaptor apoptosis-associated speck-like protein containing a CARD (ASC) to form inflammasome specks, within which the inflammatory caspase 1 is recruited and activated. Inflammasome sensors are activated in response to different triggers and differ in their overall specificities to PAMPs or DAMPs. NLRP3, the most broadly activated inflammasome sensor and a member of the nucleotide-binding domain- and leucine-rich repeat-containing protein (NLR) family, responds to an array of insults to the cell that cause cytosolic K+ efflux, Ca2+ cytosolic influx or release of mitochondrial reactive oxygen species (ROS)31,32. These insults include extracellular ATP, membrane permeabilization by pore-forming toxins and large extracellular aggregates such as uric acid crystals, cholesterol crystals and amyloids30. Other sensors, such as AIM2 and NLRC4, are tuned to recognize specific PAMPs and DAMPs, such as cytosolic double-stranded DNA and bacterial proteins, respectively31. In a parallel pathway, the mouse inflammatory caspase 11 and human caspase 4 and caspase 5 sense PAMPs and DAMPs such as bacterial lipopolysaccharide (LPS) that gain cytosolic access and endogenous oxidized phospholipids, leading directly to membrane damage or pyroptosis, and secondary K+ efflux followed by noncanonical NLRP3 inflammasome activation33–36.
For More Information: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8351223/