Authors: Meaghan E Colling 1, Yogendra Kanthi 2 PMID: 32558620
PMCID: PMC7306998 OI: 10.1177/1358863X20932640
Abstract
An ongoing global pandemic of viral pneumonia (coronavirus disease [COVID-19]), due to the virus SARS-CoV-2, has infected millions of people and remains a threat to many more. Most critically ill patients have respiratory failure and there is an international effort to understand mechanisms and predictors of disease severity. Coagulopathy, characterized by elevations in D-dimer and fibrin(ogen) degradation products (FDPs), is associated with critical illness and mortality in patients with COVID-19. Furthermore, increasing reports of microvascular and macrovascular thrombi suggest that hemostatic imbalances may contribute to the pathophysiology of SARS-CoV-2 infection. We review the laboratory and clinical findings of patients with COVID–19-associated coagulopathy, and prior studies of hemostasis in other viral infections and acute respiratory distress syndrome. We hypothesize that an imbalance between coagulation and inflammation may result in a hypercoagulable state. Although thrombosis initiated by the innate immune system is hypothesized to limit SARS-CoV-2 dissemination, aberrant activation of this system can cause endothelial injury resulting in loss of thromboprotective mechanisms, excess thrombin generation, and dysregulation of fibrinolysis and thrombosis. The role various components including neutrophils, neutrophil extracellular traps, activated platelets, microparticles, clotting factors, inflammatory cytokines, and complement play in this process remains an area of active investigation and ongoing clinical trials target these different pathways in COVID-19.Keywords anticoagulation, antiplatelet, COVID-19, inflammation, neutrophils, thrombosis, vascular endothelium, venous thromboembolism (VTE)
Introduction
In December 2019, a new betacoronavirus (severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]), thought to originate in Wuhan, China, emerged as a novel human pathogen for viral pneumonia (coronavirus disease [COVID-19]), resulting in an ongoing pandemic.1,2 The number of cases worldwide now exceeds five million, with more than 350,000 associated deaths, triggering a global effort to understand the predictors of disease severity for rapid triage, and the pathology of disease for rational therapeutic development and clinical trials. A consistent finding in early case series in China and New York City is an association between elevations in D-dimer and fibrin(ogen) degradation products (FDPs) and increasing COVID-19 severity and mortality.3–7 We aim to review the available data on the coagulopathy observed in COVID-19 and draw from studies of prior viral epidemics to explore possible mechanisms and therapies.
Coronaviruses are enveloped, non-segmented, positive-sense RNA viruses of the Nidovirales order within the Coronaviridae family. Different strains are infectious to a broad range of animals including humans, bats, cats, racoon dogs, rabbits, pigs, and cattle.8 In general, coronavirus infections in humans are mild; however, two recent epidemics of betacoronaviruses – SARS in 20039–11 and Middle East Respiratory Syndrome (MERS) in 201212,13 – were associated with significant mortality with death rates around 10% and 35%, respectively.14,15 While the observed case fatality rate for the COVID-19 pandemic is lower,16,17 the population at risk is much higher due to the global spread of the disease and the infectivity of the virus,18 and worldwide fatalities already exceed those in the prior epidemics.
Common clinical manifestations of patients with COVID-19 include fever and cough, and less commonly fatigue, dyspnea, headache, sore throat, anosmia, nausea, vomiting, or diarrhea.6 In the largest case series to date of over 44,000 patients with COVID-19, > 75% of cases were mild, 14% were severe, and 5% were critical, with an overall case fatality rate of 2–2.5%. All deaths occurred in patients with critical disease (in which the case fatality rate was almost 50%).19 While the majority of critically ill patients with COVID-19 have isolated respiratory failure, often acute respiratory distress syndrome (ARDS), multiple organ dysfunction occurs in 20–30% of patients with critical illness and more often in fatal cases.16 Hematologic findings, such as mild to moderate thrombocytopenia and lymphopenia, are associated with COVID-19;20,21 however, the most significant and concerning vascular aspect of this disease is coagulopathy. We have attempted to summarize the data on the pathogenesis, epidemiology and outcomes related to COVID-19-coagulopathy and thrombotic disease using PubMed as well as the pre-print server https://medrxiv.org (date of last search April 23, 2020).
Coagulopathy of SARS-CoV-2 and other infections
There is particular interest in the coagulopathy in patients with COVID-19 as abnormal coagulation parameters, most consistently elevations in D-dimer and FDPs, are associated with disease severity.22,23 An elevated D-dimer, the most common coagulation abnormality in COVID-19 (found in up to 45% of patients), is an independent risk factor for death,6,22,24,25 and patients with D-dimer greater than 1000 ng/mL are almost 20 times more likely to die from their infection than patients with lower D-dimer values.25 In contrast, most patients with COVID-19 have a normal or mildly prolonged prothrombin time (PT) and a normal or shortened activated partial thromboplastin time (aPTT) on presentation and these labs are not reliably associated with disease severity.5,17,22,24,25 Both initial and longitudinal monitoring of coagulation parameters can predict disease severity, as elevated D-dimer and FDP levels on admission and decreased levels of fibrinogen and antithrombin III during the admission are associated with death.23 Although changes in plasminogen activator inhibitor-1 (PAI-1) levels and activity have not been studied, an increase in the PAI-1/tissue plasminogen activator (t-PA) ratio would not be unexpected. These findings may be due to uncontrolled activation of coagulation with ongoing consumption and widespread microvascular thrombosis.
While early descriptions of the coagulopathy identified it as disseminated intravascular coagulation (DIC), in DIC, unlike in severe COVID-19, platelet count and PT prolongation correlate with sepsis severity and mortality, while fibrinogen and FDPs levels do not.26,27 And while the majority of patients who die from COVID-19 develop some laboratory evidence of DIC during their admission, elevations in D-dimer and prolonged PT with mild thrombocytopenia and normal fibrinogen are commonly seen.23 Thromboelastography in patients with COVID-19 in the ICU shows a hypercoagulable state.28 These observations suggest the underlying pathophysiology in at least a subset of critically ill patients with COVID-19 is distinct from traditional systemic DIC and may be due to a unique coagulopathy.
Elevations in D-dimer are common in critical illness and are associated with disease severity and mortality in many severe infections.29–31 Patients with influenza, SARS, HIV, hantavirus, Ebola virus, and dengue have elevations in D-dimer, prothrombin fragments, thrombin–antithrombin complexes, and/or plasmin-α2-antiplasmin complexes.32 Similar to patients with SARS-CoV-2 infections, there is an association between elevated D-dimer and mortality in patients with H1N1 and H5N1, which is not seen in SARS.33–35
Additionally, in the H1N1 pandemic, patients with severe disease had high rates of venous thromboembolism (VTE) and many patients with thromboembolism did not have evidence of systemic DIC.36–39 Patients with ARDS from H1N1 infection had a greater than 20-fold increase in risk of pulmonary embolism compared to patients with ARDS unrelated to H1N1.39 Empiric therapeutic anticoagulation in patients with ARDS was associated with decreased rates of VTE in patients with ARDS from H1N1, but had no effect on VTE rates in patients with ARDS unrelated to H1N1 infection. There are reports of VTE in patients with COVID-19, despite concerns regarding underdiagnosis given baseline elevations in D-dimer, as well as pragmatic challenges in diagnostic imaging while in isolation, including use of personal protective equipment and longer duration of exposure of health care workers.40,41 Although data remain scarce, there are increasing reports of arterial thrombotic events including ischemic strokes in patients with COVID-19.41–43 Myocardial injury, defined by elevations in cardiac troponin levels, is common in patients hospitalized with COVID-19 and is associated with severe disease and high risk of mortality.44,45 Myocardial injury may result from systemic inflammatory response syndrome (SIRS) and inflammation as well as due to acute thrombotic events.46,47 Similar observations of myocardial injury have been found in patients with other viral infections.48,49
Pathologic findings in SARS-CoV-2 infection
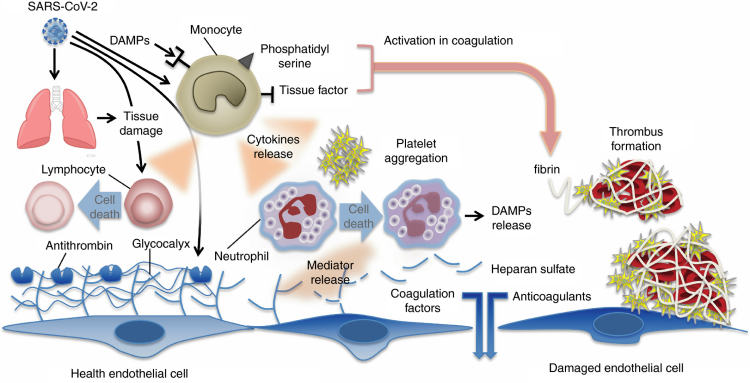
Although there are only a few published pathologic reports of patients with COVID-19, histopathology of lung specimens from patients with early disease shows characteristic findings of ARDS and evidence of small vessel occlusion.50,51 There are several mechanisms by which SARS-CoV-2 infection may result in microvascular and macrovascular thrombosis, including cytokine storm with activation of leukocytes, endothelium and platelets resulting in upregulation of tissue factor, activation of coagulation, thrombin generation and fibrin formation,52 deranged coagulation with imbalances in PAI-1, tissue factor pathway inhibitor, and activated protein C that promotes fibrin generation and limits fibrinolysis,53,54 hypoxic vaso-occlusion, and direct viral effects with cell activation (Figure 1). It remains an active area of investigation whether these are specific to SARS-CoV-2 infection or a final common pathway in the thromboinflammatory response to viral infections and a marker of disease severity. Early COVID-19 autopsy reports have also identified a possible role for neutrophils as microvascular thrombi contained numerous neutrophils, which in some cases were partially degenerated, consistent with neutrophil extracellular traps (NETs).55,56 NETs are tangles of DNA released from neutrophils, and are decorated with antimicrobial and nuclear proteins that propagate intravascular thrombosis.57,58 NETs initiate both the extrinsic and contact pathways by augmenting presentation of tissue factor, activation of factor XII (FXII), as well as trapping and activating platelets.59–62 Consistent with these observations, patients with severe COVID-19 have elevated serum markers of neutrophil activation and NET formation.63 In one study, neutrophil activation measured in serum correlated with, and sometimes preceded, VTE in patients with COVID-19.64
Figure 1. Immune activation and mechanisms of coagulopathy in patients with coronavirus disease 2019 (COVID-19).
Multiple processes may contribute to COVID-19-associated coagulopathy including direct infection of type II pneumocytes and endothelial cells, leading to barrier dysfunction and increased permeability; inflammatory responses characterized by activation of T cells, neutrophils, monocytes, macrophages, and platelets resulting in exuberant inflammatory cytokine release (including IL-1, IL-6, IL-10, TNF-α), monocyte-derived TF and PAI-1 expression; and culminating in the development of microvascular and macrovascular thrombi composed of fibrin, NETs, and platelets.
IL, interleukin; NETs, neutrophil extracellular traps; PAI-1, plasminogen activator inhibitor-1; TF, tissue factor; TNF-α, tumor necrosis factor-alpha.
Dysregulation of hemostasis and coagulopathy in acute respiratory distress syndrome (ARDS)
Thrombi in the pulmonary micro- and macrovasculature are observed in patients with ARDS with or without overt DIC, and changes consistent with a prothrombotic state have been found both in blood and in alveolar fluid studies of these patients.65,66 Higher levels of FDPs and D-dimer are seen in patients who developed ARDS as compared to patients with similar predisposing conditions that did not develop ARDS.67 Lower levels of protein C and higher levels of soluble thrombomodulin and PAI-1 are also associated with multiple organ failure, disease severity, and mortality in ARDS in some studies.53,68–72 Finally, plasma and alveolar levels of tissue factor are higher in patients with ARDS than patients with pulmonary edema.73 Mechanistically, there is increased thrombin generation by tissue factor coupled with an impaired fibrinolytic response due to elevations in PAI-1. Elevations in D-dimer, a breakdown product of crosslinked fibrin, may result from residual t-PA/plasmin activity, as well as from alternative fibrinolytic pathways such as human neutrophil elastase activity.74,75
As patients with COVID-19 frequently have isolated pulmonary findings, the initial hemostatic dysregulation may be localized to the lungs as a consequence of the bidirectional relationship between the innate immune system and thrombosis. Activated platelets through degranulation and coordinated interactions with monocytes, dendritic cells, and neutrophils, as well as activated T cells, NETs, tissue factor-bearing microparticles, and coagulation proteases may facilitate this crosstalk.54,76,77 In this model, immune cells, inflammatory cytokines, and pathogen-associated molecular patterns induce thrombi consisting of fibrin, monocytes, neutrophils, and platelets.57,58,78 These immunothrombi initially serve a protective purpose, promoting pathogen recognition and creating a sterile barrier against further pathogen invasion, but can become maladaptive and injurious to tissue and organ perfusion.57,79,80 During this process, there is abundant intra- and extra-vascular fibrin deposition and impaired fibrinolysis, which has been well described in ARDS.81,82 In postmortem studies, both macro- and microvascular thrombi are common in patients in ARDS (observed in up to 95% of patients).82,83 In COVID-19, the alveolar immunothrombotic response may be an attempt to limit dissemination of SARS-CoV-2 outside the alveoli.
Findings from the SARS epidemic provide possible viral-specific mechanisms for ARDS and uncontrolled coagulation. Autopsy studies of patients who died of SARS pneumonia, identified the SARS-CoV spike (S) protein in cells expressing the receptor angiotensin-converting enzyme 2 (ACE2),84–87 the leading candidate receptor for SARS-CoV-2.88,89 Binding of the S protein to ACE2 induces expression of a nuclear factor kappa B (NFκB)-driven inflammatory module, resulting in production of proinflammatory cytokines including monocyte chemoattractant protein 1 (MCP-1), transforming growth factor-beta 1 (TGF-β1), tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, and IL-6, which have been implicated in thrombogenesis.90 Although inflammatory responses are important in host-defense, hyperinflammatory responses result in tissue damage, disruption of the endothelial barrier, and uncontrolled activation of coagulation.54 Overall, these findings are consistent with a model in which SARS-CoV and SARS-CoV-2 directly infect endothelial and epithelial cells, increasing levels of proinflammatory cytokines, causing immune-mediated damage to the vasculature and surrounding tissue, with exposure of tissue factor and associated thromboinflammatory changes.91 While these changes appear to be predominantly in the lungs, endotheliitis in COVID-19 has been observed in kidneys, liver, heart, and intestine.91
Additional studies in SARS-CoV and influenza found dysregulation of urokinase, coagulation, and fibrinolysis pathways contributed to the severity of lung injury, possibly through altering the hemostatic balance with subsequent coagulation-induced ischemic injury.92 Plasminogen was protective against severe influenza A, H5N1, and H1N1 infections.93 These groups hypothesized that increased fibrinolysis led to a positive feedback loop of vascular permeability, leukocyte recruitment, and fibrin generation. Interestingly, one hypothesis suggests that elevated plasminogen may be a risk factor for SARS-CoV-2 infection because plasmin may cleave the S protein of the virus and increase its infectivity.94 These findings highlight the delicate balance between corralling infection and uncontrolled inflammation and thrombosis.
Therapeutic considerations
Markers of hypercoagulability and higher inflammatory mediators are consistently associated with worse outcomes in patients with ARDS and sepsis. These observations have led to numerous clinical trials targeting various components of inflammatory and coagulation pathways in acute lung injury, ARDS or sepsis. Studies with heparin, steroids, non-steroidal anti-inflammatory drugs, and TNF-α inhibitors have been disappointing.95–100
Given the laboratory and clinical findings in patients with severe COVID-19, several repurposed and novel therapies are under investigation in clinical trials to prevent the hyperinflammatory response or mitigate uncontrolled coagulation. As elevations in D-dimer and FDPs likely reflect ongoing lung injury and microvascular thrombi, possible therapeutic targets include inflammatory cytokines, activated platelets, neutrophils, or microparticles that may propagate thrombosis; or anticoagulants and fibrinolytics that could limit thrombosis. Supporting this enthusiasm was a recent retrospective study in China in which VTE prophylaxic dose heparin was associated with a survival benefit in patients with severe COVID-19 and evidence of sepsis-induced coagulopathy.101 The study found no benefit among patients with milder COVID-19 illness; however, the study did not control for other markers of disease severity nor other therapies, such as antivirals. The study raises the possibility that prophylactic or therapeutic anticoagulation may benefit patients with severe infection. Heparin may alter the biology of the disease not only through its anticoagulant properties, but also due to its anti-inflammatory effects that promote a quiescent endothelium.
Current expert recommendations, including interim guidelines from the International Society on Thrombosis and Haemostasis (ISTH) and the American College of Cardiology (ACC), recommend use of prophylactic dose LMWH or unfractionated heparin in all COVID-19 patients requiring hospital admission; for patients with a contraindication to pharmacologic prophylaxis, mechanical prophylaxis should be used.102,103 While a number of VTE risk stratification tools exist for hospitalized medical patients, these have not been validated in patients with COVID-19. Extended VTE prophylaxis with LMWH or direct oral anticoagulants after hospitalization for acute medical illness reduces the risk of VTE with an associated increased risk of bleeding.104–106 There are currently no data regarding extended prophylaxis in patients with COVID-19; however, the ACC expert opinion statement recommends consideration of extended prophylaxis in patients with elevated risk of VTE, such as patients with cancer or prolonged immobility who have low bleeding risk. Given early reports and ongoing concerns of high rates of VTE, randomized trials of empiric therapeutic anticoagulation or antifibrinolytics are ongoing, and there are reports of empiric therapeutic anticoagulation in patients with significantly elevated D-dimer both in Italy and in the US. While heparin offers both anti-inflammatory and anticoagulant effects, the benefit of therapeutic anticoagulation remains uncertain, with a risk of bleeding complications in critically ill patients with respiratory failure.95,107 Clinical trials will help define the role of heparin in the treatment of hospitalized patients with COVID-19. Outside of a trial setting, we advocate universal standard-dose pharmacologic VTE prophylaxis in patients without a contraindication. In patients with a high suspicion of VTE where access to confirmatory or serial imaging is limited, clinicians may consider empiric anticoagulation, although there is a paucity of evidence to provide guidance in this context. There are currently no randomized data to recommend empiric therapeutic or intermediate-dose anticoagulation in patients without documented VTE, or an other indication for anticoagulation, or outside the context of a clinical trial. A recent retrospective, observational study in New York City showed therapeutic anticoagulation was associated with decreased mortality in patients with COVID-19 who required mechanical ventilation, but not in all hospitalized patients with COVID-19. Although these findings are provocative, interpretation is limited by their observational nature.108
There are over 300 trials ongoing for patients with COVID-19, many of which aim to simultaneously reduce inflammation and thrombosis, including cytokine-directed therapies (against IL-1, IL-6, interferon gamma), corticosteroids, Janus kinase inhibitors, TLR ligands, complement inhibitors, N-acetylcysteine, serine protease inhibitors, DNAse enzymes, and anti-viral agents. However, suppressing the cytokine storm or hypercoagulability may be insufficient once initiated, and targeting upstream pathways to prevent activation of this self-amplifying feedback loop may be more effective.
One therapeutic candidate to treat COVID-19 is dipyridamole, an adenosinergic drug indicated for use as an arterial thromboembolic prophylaxis agent in combination with aspirin or warfarin.109 Dipyridamole has recently been shown to suppress human neutrophil and T-cell activation, upstream of cytokine effectors.58,110 Dipyridamole induces a type I interferon response, which is necessary for physiologic anti-viral activity, and inhibits SARS-CoV-2 replication in vitro by inhibiting a critical viral replication complex.111,112 Administered orally, dipyridamole has a favorable safety profile, and a small clinical trial in patients with COVID-19 suggests it may improve D-dimer levels.113 Randomized clinical trials of agents active at the intersection of inflammation and coagulation in COVID-19, such as dipyridamole, t-PA, and heparin are necessary to determine if these therapeutics can restore the balance of inflammation and coagulation without dampening early or late physiologic anti-viral responses. The heterogenous response to the SARS-CoV-2 infection and the various time-dependent pathways driving pathology make universal therapies challenging. The temporal and mechanistic role each pathway plays in severe SARS-CoV-2 infection remains uncertain and requires further exploration for treatment opportunities as efforts to control this pandemic continue.
Conclusions
In conclusion, in patients with COVID-19, the presence of coagulopathy, characterized by elevations in D-dimer and FDPs, is consistently associated with more severe illness and mortality. Laboratory, clinical, and early histopathologic findings suggest this coagulopathy is distinct from sepsis-induced DIC and may reflect dysregulated hemostasis. Similar findings have been associated with several other viral infections, and it remains uncertain if this coagulopathy is specific to SARS-CoV-2 or the end common pathway of the thrombo-inflammatory response to severe viral infections. There are efforts to target numerous components of the thrombo-inflammatory pathway to improve outcomes in patients with severe COVID-19. The optimal management for these patients including strategies to diagnose VTE, appropriate anticoagulation doses and duration, and effectiveness of novel therapies are under active investigation in the current pandemic.
Acknowledgements
The authors would like to thank Charles Bolan, MD and Jason Knight, MD, PhD for guidance and review of the manuscript, and all members of the ‘NETwork to Target Neutrophils in COVID-19’ and the SVM Next Generation Committee for their helpful advice and encouragement. The authors credit Alan Hoofring for the illustration.
Declaration of conflicting interests
Yogen Kanthi has served as a consultant for Surface Oncology and has a pending patent on use of biogases in vascular disease. Meaghan E. Colling has nothing to disclose.
Funding
Meaghan E. Colling is supported by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health (NIH). Yogen Kanthi is supported by grant funding from the NIH-NHLBI (K08HL131993, R01HL150392), A. Alfred Taubman Medical Research Institute, Michigan Medicine Frankel COVID-19 Cardiovascular Impact Research Ignitor Program, Falk Medical Research Trust Catalyst Award, American Venous Forum-JOBST Award, University of Michigan BioInterfaces Institute, and Bo Schembechler Heart of A Champion Foundation.
ORCID iD
Yogendra Kanthi https://orcid.org/0000-0002-5660-5194
References
| 1. | World Health Organization . Pneumonia of unknown cause – China. Disease outbreak news, 5 January, https://www.who.int/csr/don/05-january-2020-pneumonia-of-unkown-cause-china/en/ (2020, accessed 25 March 2020). Google Scholar |
| 2. | Zhu, N, Zhang, D, Wang, W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020; 382: 727–733. Google Scholar | Crossref | Medline |
| 3. | Gao, Y, Li, T, Han, M, et al. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J Med Virol. 2020; 92: 791–796. Google Scholar | Crossref | Medline |
| 4. | Wan, S, Xiang, Y, Fang, W, et al. Clinical features and treatment of COVID-19 patients in northeast Chongqing. J Med Virol. 2020; 92: 797–806. Google Scholar | Crossref | Medline |
| 5. | Huang, C, Wang, Y, Li, X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020; 395: 497–506. Google Scholar | Crossref | Medline |
| 6. | Guan, WJ, Ni, ZY, Hu, Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020; 382: 1708–1720. Google Scholar | Crossref | Medline |
| 7. | Petrilli, CM, Jones, SA, Yang, J, et al. Factors associated with hospitalization and critical illness among 4,103 patients with COVID-19 disease in New York City. BMJ 2020; 369: m1966. Google Scholar | Crossref | Medline |
| 8. | Saif, LJ. Animal coronavirus vaccines: lessons for SARS. Dev Biol (Basel) 2004; 119: 129–140. Google Scholar | Medline |
| 9. | Kuiken, T, Fouchier, RA, Schutten, M, et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003; 362: 263–270. Google Scholar | Crossref | Medline | ISI |
| 10. | Drosten, C, Gunther, S, Preiser, W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1967–1976. Google Scholar | Crossref | Medline | ISI |
| 11. | Ksiazek, TG, Erdman, D, Goldsmith, CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003; 348: 1953–1966. Google Scholar | Crossref | Medline | ISI |
| 12. | De Groot, RJ, Baker, SC, Baric, RS, et al. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J Virol 2013; 87: 7790–7792. Google Scholar | Crossref | Medline | ISI |
| 13. | Zaki, AM, van Boheemen, S, Bestebroer, TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367: 1814–1820. Google Scholar | Crossref | Medline | ISI |
| 14. | World Health Organization . Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003, https://www.who.int/csr/sars/country/table2004_04_21/en/ (2003, accessed 28 March 2020). Google Scholar |
| 15. | World Health Organization . Middle East respiratory syndrome coronavirus (MERS-CoV). MERS Monthly Summary, November 2019, http://www.who.int/emergencies/mers-cov/en/ (2019, accessed 27 March 2020). Google Scholar |
| 16. | Yang, X, Yu, Y, Xu, J, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir Med 2020; 8: 475–481. Google Scholar | Crossref | Medline |
| 17. | Wang, D, Hu, B, Hu, C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 2020; 323: 1061–1069. Google Scholar | Crossref | Medline |
| 18. | Petrosillo, N, Viceconte, G, Ergonul, O, et al. COVID-19, SARS and MERS: Are they closely related? Clin Microbiol Infect 2020; 26:729–734. Google Scholar | Crossref | Medline |
| 19. | Wu, Z, McGoogan, JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020; 323: 1239–1242. Google Scholar | Crossref | Medline |
| 20. | Zhou, P, Yang, XL, Wang, XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; 579: 270–273. Google Scholar | Crossref | Medline |
| 21. | Lippi, G, Plebani, M, Henry, BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin Chim Acta 2020; 506: 145–148. Google Scholar | Crossref | Medline |
| 22. | Wu, C, Chen, X, Cai, Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med 2020; 180: 1–11. Google Scholar | Crossref |
| 23. | Tang, N, Li, D, Wang, X, et al. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 2020; 18: 844–847. Google Scholar | Crossref | Medline |
| 24. | Chen, N, Zhou, M, Dong, X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020; 395: 507–513. Google Scholar | Crossref | Medline |
| 25. | Zhou, F, Yu, T, Du, R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020; 395: 1054–1062. Google Scholar | Crossref | Medline |
| 26. | Iba, T, Levy, JH, Warkentin, TE, et al. Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Thromb Haemost 2019; 17: 1989–1994. Google Scholar | Crossref | Medline |
| 27. | Iba, T, Di Nisio, M, Thachil, J, et al. A proposal of the modification of Japanese Society on Thrombosis and Hemostasis (JSTH) Disseminated Intravascular Coagulation (DIC) diagnostic criteria for sepsis-associated DIC. Clin Appl Thromb Hemost 2018; 24: 439–445. Google Scholar | SAGE Journals |
| 28. | Panigada, M, Bottino, N, Tagliabue, P, et al. Hypercoagulability of COVID-19 patients in intensive care unit. A report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost 2020; 18: 1738–1742. Google Scholar | Crossref | Medline |
| 29. | Shorr, AF, Thomas, SJ, Alkins, SA, et al. D-dimer correlates with proinflammatory cytokine levels and outcomes in critically ill patients. Chest 2002; 121: 1262–1268. Google Scholar | Crossref | Medline | ISI |
| 30. | Rodelo, JR, De la Rosa, G, Valencia, ML, et al. D-dimer is a significant prognostic factor in patients with suspected infection and sepsis. Am J Emerg Med 2012; 30: 1991–1999. Google Scholar | Crossref | Medline |
| 31. | Wan, J, Yang, X, He, W, et al. Serum D-dimer levels at admission for prediction of outcomes in acute pancreatitis. BMC Gastroenterol 2019; 19: 67. Google Scholar | Crossref | Medline |
| 32. | Goeijenbier, M, van Wissen, M, van de Weg, C, et al. Review: Viral infections and mechanisms of thrombosis and bleeding. J Med Virol 2012; 84: 1680–1696. Google Scholar | Crossref | Medline |
| 33. | Wong, RS, Wu, A, To, KF, et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003; 326: 1358–1362. Google Scholar | Crossref | Medline |
| 34. | Soepandi, PZ, Burhan, E, Mangunnegoro, H, et al. Clinical course of avian influenza A(H5N1) in patients at the Persahabatan Hospital, Jakarta, Indonesia, 2005–2008. Chest 2010; 138: 665–673. Google Scholar | Crossref | Medline |
| 35. | Wang, ZF, Su, F, Lin, XJ, et al. Serum D-dimer changes and prognostic implication in 2009 novel influenza A(H1N1). Thromb Res 2011; 127: 198–201. Google Scholar | Crossref | Medline |
| 36. | Centers for Disease Control and Prevention . Intensive-care patients with severe novel influenza A (H1N1) virus infection – Michigan, June 2009. MMWR Morb Mortal Wkly Rep 2009; 58: 749–752. Google Scholar | Medline |
| 37. | Avnon, LS, Munteanu, D, Smoliakov, A, et al. Thromboembolic events in patients with severe pandemic influenza A/H1N1. Eur J Intern Med 2015; 26: 596–598. Google Scholar | Crossref | Medline |
| 38. | Bunce, PE, High, SM, Nadjafi, M, et al. Pandemic H1N1 influenza infection and vascular thrombosis. Clin Infect Dis 2011; 52: e14–17. Google Scholar | Crossref | Medline |
| 39. | Obi, AT, Tignanelli, CJ, Jacobs, BN, et al. Empirical systemic anticoagulation is associated with decreased venous thromboembolism in critically ill influenza A H1N1 acute respiratory distress syndrome patients. J Vasc Surg Venous Lymphat Disord 2019; 7: 317–324. Google Scholar | Crossref | Medline |
| 40. | Cui, S, Chen, S, Li, X, et al. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost. Epub ahead of print 6 May 2020. DOI: 10.1111/jth.14830. Google Scholar | Crossref |
| 41. | Klok, FA, Kruip, M, van der Meer, NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 2020; 191: 141–147. Google Scholar |
| 42. | Lodigiani, C, Iapichino, G, Carenzo, L, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res 2020; 191: 9–14. Google Scholar | Crossref | Medline |
| 43. | Oxley, TJ, Mocco, J, Majidi, S, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med 2020; 382: e60. Google Scholar | Crossref | Medline |
| 44. | Guo, T, Fan, Y, Chen, M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol 2020; 5: 811–818. Google Scholar | Crossref | Medline |
| 45. | Shi, S, Qin, M, Shen, B, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol 2020; 5: 802–810. Google Scholar | Crossref | Medline |
| 46. | Lacour, T, Semaan, C, Genet, T, et al. Insights for increased risk of failed fibrinolytic therapy and stent thrombosis associated with COVID-19 in ST-segment elevation myocardial infarction patients. Catheter Cardiovasc Interv. Epub ahead of print 30 April 2020. DOI: 10.1002/ccd.28948. Google Scholar | Crossref |
| 47. | Corrales-Medina, VF, Madjid, M, Musher, DM. Role of acute infection in triggering acute coronary syndromes. Lancet Infect Dis 2010; 10: 83–92. Google Scholar | Crossref | Medline | ISI |
| 48. | Madjid, M, Aboshady, I, Awan, I, et al. Influenza and cardiovascular disease: Is there a causal relationship? Tex Heart Inst J 2004; 31: 4–13. Google Scholar | Medline | ISI |
| 49. | Kwong, JC, Schwartz, KL, Campitelli, MA, et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N Engl J Med 2018; 378: 345–353. Google Scholar | Crossref | Medline |
| 50. | Tian, S, Hu, W, Niu, L, et al. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol 2020; 15: 700–704. Google Scholar | Crossref | Medline |
| 51. | Xu, Z, Shi, L, Wang, Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 2020; 8: 420–422. Google Scholar | Crossref | Medline |
| 52. | Sebag, SC, Bastarache, JA, Ware, LB. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr Pharm Biotechnol 2011; 12: 1481–1496. Google Scholar | Crossref | Medline |
| 53. | Ware, LB, Fang, X, Matthay, MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol 2003; 285: L514–521. Google Scholar | Crossref | Medline | ISI |
| 54. | Frantzeskaki, F, Armaganidis, A, Orfanos, SE. Immunothrombosis in acute respiratory distress syndrome: Cross talks between inflammation and coagulation. Respiration 2017; 93: 212–225. Google Scholar | Crossref | Medline |
| 55. | Fox, SE, Akmatbekov, A, Harbert, JL, et al. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir Med 2020; 8: 681–686. Google Scholar | Crossref | Medline |
| 56. | Barnes, BJ, Adrover, JM, Baxter-Stoltzfus, A, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med 2020; 217: e20200652. Google Scholar | Crossref | Medline |
| 57. | Yadav, V, Chi, L, Zhao, R, et al. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1beta-driven venous thrombosis. J Clin Invest 2019; 129: 2872–2877. Google Scholar | Crossref | Medline |
| 58. | Ali, RA, Gandhi, AA, Meng, H, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun 2019; 10: 1916. Google Scholar | Crossref |
| 59. | Kambas, K, Mitroulis, I, Ritis, K. The emerging role of neutrophils in thrombosis—The journey of TF through NETs. Front Immunol 2012; 3: 385. Google Scholar | Crossref | Medline |
| 60. | Liberale, L, Holy, EW, Akhmedov, A, et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. J Clin Med 2019; 8: 2072. Google Scholar | Crossref |
| 61. | Noubouossie, DF, Reeves, BN, Strahl, BD, et al. Neutrophils: Back in the thrombosis spotlight. Blood 2019; 133: 2186–2197. Google Scholar | Crossref | Medline |
| 62. | Thalin, C, Hisada, Y, Lundstrom, S, et al. Neutrophil extracellular traps: Villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol 2019; 39: 1724–1738. Google Scholar | Crossref | Medline |
| 63. | Zuo, Y, Yalavarthi, S, Shi, H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. Epub ahead of print 24 April 2020. DOI: 10.1172/jci.insight.138999. Google Scholar | Crossref |
| 64. | Zuo, Y, Zuo, M, Yalavarthi, S, et al. Neutrophil extracellular traps and thrombosis in COVID-19. medRxiv. Preprint 5 May 2020. DOI: 10.1101/2020.04.30.20086736. Google Scholar | Crossref |
| 65. | Bone, RC, Francis, PB, Pierce, AK. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med 1976; 61: 585–589. Google Scholar | Crossref | Medline | ISI |
| 66. | Blondonnet, R, Constantin, JM, Sapin, V, et al. A pathophysiologic approach to biomarkers in acute respiratory distress syndrome. Dis Markers 2016; 2016: 3501373. Google Scholar | Crossref | Medline |
| 67. | Haynes, JB, Hyers, TM, Giclas, PC, et al. Elevated fibrin(ogen) degradation products in the adult respiratory distress syndrome. Am Rev Respir Dis 1980; 122: 841–847. Google Scholar | Medline |
| 68. | Sapru, A, Calfee, CS, Liu, KD, et al. Plasma soluble thrombomodulin levels are associated with mortality in the acute respiratory distress syndrome. Int Care Med 2015; 41: 470–478. Google Scholar | Crossref | Medline |
| 69. | Ware, LB, Matthay, MA, Parsons, PE, et al. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit Care Med 2007; 35: 1821–1828. Google Scholar | Medline | ISI |
| 70. | Thompson, BT, Chambers, RC, Liu, KD. Acute respiratory distress syndrome. N Engl J Med 2017; 377: 1904–1905. Google Scholar | Crossref | Medline |
| 71. | Prabhakaran, P, Ware, LB, White, KE, et al. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol 2003; 285: L20–28. Google Scholar | Crossref | Medline |
| 72. | Agrawal, A, Zhuo, H, Brady, S, et al. Pathogenetic and predictive value of biomarkers in patients with ALI and lower severity of illness: Results from two clinical trials. Am J Physiol Lung Cell Mol Physiol 2012; 303: L634–639. Google Scholar | Crossref | Medline |
| 73. | Bastarache, JA, Wang, L, Geiser, T, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax 2007; 62: 608–616. Google Scholar | Crossref | Medline |
| 74. | Bach-Gansmo, ET, Halvorsen, S, Godal, HC, et al. D-dimers are degraded by human neutrophil elastase. Thromb Res 1996; 82: 177–186. Google Scholar | Crossref | Medline |
| 75. | Gando, S, Hayakawa, M, Sawamura, A, et al. The activation of neutrophil elastase-mediated fibrinolysis is not sufficient to overcome the fibrinolytic shutdown of disseminated intravascular coagulation associated with systemic inflammation. Thromb Res 2007; 121: 67–73. Google Scholar | Crossref | Medline |
| 76. | Koupenova, M, Clancy, L, Corkrey, HA, et al. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res 2018; 122: 337–351. Google Scholar | Crossref | Medline |
| 77. | Mackman, N. The many faces of tissue factor. J Thromb Haemost 2009; 7(suppl 1): 136–139. Google Scholar | Crossref | Medline |
| 78. | Chang, JC. Acute respiratory distress syndrome as an organ phenotype of vascular microthrombotic disease: Based on hemostatic theory and endothelial molecular pathogenesis. Clin Appl Thromb Hemost 2019; 25: 1076029619887437. Google Scholar | SAGE Journals |
| 79. | Van der Poll, T, Herwald, H. The coagulation system and its function in early immune defense. Thromb Haemost 2014; 112: 640–648. Google Scholar | Crossref | Medline |
| 80. | Lefrancais, E, Mallavia, B, Zhuo, H, et al. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018; 3: e98178. Google Scholar | Crossref | Medline |
| 81. | Glas, GJ, Van Der Sluijs, KF, Schultz, MJ, et al. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J Thromb Haemost 2013; 11: 17–25. Google Scholar | Crossref | Medline |
| 82. | Tomashefski, JF Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med 2000; 21: 435–466. Google Scholar | Crossref | Medline | ISI |
| 83. | Vesconi, S, Rossi, GP, Pesenti, A, et al. Pulmonary microthrombosis in severe adult respiratory distress syndrome. Crit Care Med 1988; 16: 111–113. Google Scholar | Crossref | Medline | ISI |
| 84. | He, Y, Zhou, Y, Liu, S, et al. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem Biophys Res Commun 2004; 324: 773–781. Google Scholar | Crossref | Medline |
| 85. | Li, W, Greenough, TC, Moore, MJ, et al. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin-converting enzyme 2. J Virol 2004; 78: 11429–11433. Google Scholar | Crossref | Medline |
| 86. | Li, W, Moore, MJ, Vasilieva, N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426: 450–454. Google Scholar | Crossref | Medline | ISI |
| 87. | Xiao, X, Chakraborti, S, Dimitrov, AS, et al. The SARS-CoV S glycoprotein: Expression and functional characterization. Biochem Biophys Res Commun 2003; 312: 1159–1164. Google Scholar | Crossref | Medline | ISI |
| 88. | Hoffmann, M, Kleine-Weber, H, Schroeder, S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020; 181: 271–280.e8. Google Scholar | Crossref | Medline |
| 89. | Wrapp, D, Wang, N, Corbett, KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020; 367: 1260–1263. Google Scholar | Crossref | Medline |
| 90. | He, L, Ding, Y, Zhang, Q, et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J Pathol 2006; 210: 288–297. Google Scholar | Crossref | Medline | ISI |
| 91. | Varga, Z, Flammer, AJ, Steiger, P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020; 395: 1417–1418. Google Scholar | Crossref | Medline |
| 92. | Gralinski, LE, Bankhead, A, Jeng, S, et al. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio 2013; 4: e00271-13. Google Scholar | Crossref | Medline |
| 93. | Berri, F, Rimmelzwaan, GF, Hanss, M, et al. Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog 2013; 9: e1003229. Google Scholar | Crossref | Medline |
| 94. | Ji, HL, Zhao, R, Matalon, S, et al. Elevated plasmin(ogen) as a common risk factor for COVID-19 susceptibility. Physiol Rev 2020; 100: 1065–1075. Google Scholar | Crossref | Medline |
| 95. | Jaimes, F, De La Rosa, G, Morales, C, et al. Unfractioned heparin for treatment of sepsis: A randomized clinical trial (The HETRASE Study). Crit Care Med 2009; 37: 1185–1196. Google Scholar | Crossref | Medline |
| 96. | Abraham, E, Anzueto, A, Gutierrez, G, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet 1998; 351: 929–933. Google Scholar | Crossref | Medline | ISI |
| 97. | Abraham, E, Wunderink, R, Silverman, H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA 1995; 273: 934–941. Google Scholar | Crossref | Medline | ISI |
| 98. | National Heart, Lung, and Blood Institute ARDS Clinical Trials Network , Truwit, JD, Bernard, GR, et al. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med 2014; 370: 2191–2200. Google Scholar | Crossref | Medline | ISI |
| 99. | Bernard, GR, Wheeler, AP, Russell, JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group. N Engl J Med 1997; 336: 912–918. Google Scholar | Crossref | Medline | ISI |
| 100. | Steinberg, KP, Hudson, LD, Goodman, RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 2006; 354: 1671–1684. Google Scholar | Crossref | Medline | ISI |
| 101. | Tang, N, Bai, H, Chen, X, et al. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020; 18: 1094–1099. Google Scholar | Crossref | Medline |
| 102. | Thachil, J, Tang, N, Gando, S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost 2020; 18: 1023–1026. Google Scholar | Crossref | Medline |
| 103. | Bikdeli, B, Madhavan, MV, Jimenez, D, et al. COVID-19 and thrombotic or thromboembolic disease: Implications for prevention, antithrombotic therapy, and follow-up. J Am Coll Cardiol 2020; S0735-1097(20): 35008-7. Google Scholar | Crossref |
| 104. | Cohen, AT, Harrington, RA, Goldhaber, SZ, et al. Extended thromboprophylaxis with betrixaban in acutely ill medical patients. N Engl J Med 2016; 375: 534–544. Google Scholar | Crossref | Medline | ISI |
| 105. | Cohen, AT, Spiro, TE, Spyropoulos, AC; MAGELLAN Steering Committee . Rivaroxaban for thromboprophylaxis in acutely ill medical patients. N Engl J Med 2013; 368: 1945–1946. Google Scholar | Crossref | Medline |
| 106. | Dentali, F, Mumoli, N, Prisco, D, et al. Efficacy and safety of extended thromboprophylaxis for medically ill patients. A meta-analysis of randomised controlled trials. Thromb Haemost 2017; 117: 606–617. Google Scholar | Crossref | Medline |
| 107. | Cook, DJ, Fuller, HD, Guyatt, GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330: 377–381. Google Scholar | Crossref | Medline | ISI |
| 108. | Paranjpe, I, Fuster, V, Lala, A, et al. Association of treatment dose anticoagulation with in-hospital survival among hospitalized patients with COVID-19. J Am Coll Cardiol 2020; S0735-1097(20): 35218-9. Google Scholar | Crossref |
| 109. | Persantine (dipyridamole) [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc. December 2019. Google Scholar |
| 110. | Macatangay, BJC, Jackson, EK, Abebe, KZ, et al. A randomized, placebo-controlled, pilot clinical trial of dipyridamole to decrease HIV-associated chronic inflammation. J Infect Dis 2020; 221: 1598–1606. Google Scholar | Crossref | Medline |
| 111. | Li, Z, Li, X, Huang, Y-Y, et al. FEP-based screening prompts drug repositioning against COVID-19. bioRxiv. Preprint 25 March 2020. DOI: https://doi.org/10.1101/2020.03.23.004580. Google Scholar |
| 112. | Galabov, AS, Mastikova, M. Dipyridamole induces interferon in man. Biomed Pharmacother. 1984; 38: 412–413. Google Scholar | Medline |
| 113. | Liu, X, Li, Z, Liu, S, et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm Sin B 2020; 10: 1205–1215. Google Scholar | Crossref | Medline |