Authors: Toshiaki Iba 1, Jean Marie Connors 2, Jerrold H Levy 3
- PMID: 32918567 PMCID: PMC7486586 DOI: 10.1007/s00011-020-01401-6
Abstract
Background
COVID-19-associated coagulopathy (CAC) characterized by the elevated D-dimer without remarkable changes of other global coagulation markers is associated with various thrombotic complications and disease severity. The purpose of this review is to elucidate the pathophysiology of this unique coagulopathy.
Methods
The authors performed online search of published medical literature through PubMed using the MeSH (Medical Subject Headings) term “COVID-19,” “SARS-CoV-2,” “coronavirus,” “coagulopathy,” and “thrombus.” Then, selected 51 articles that closely relevant to coagulopathy in COVID-19.
Results
The primary targets of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are the pneumocytes, immune cells, and vascular endothelial cells. The alveolar damage and the pulmonary microvascular thrombosis are the major causes of acute lung injury in COVID-19. The endotheliopathy that occurs is due to direct SARS-CoV-2 infection and activation of other pathways that include the immune system and thromboinflammatory responses leading to what is termed CAC. As a result, both microvascular and macrovascular thrombotic events occur in arterial, capillary, venule, and large vein vascular beds to produce multiorgan dysfunction and thrombotic complications. In addition to the endothelial damage, SARS-CoV-2 also can cause vasculitis and presents as a systemic inflammatory vascular disease. Clinical management of COVID-19 includes anticoagulation but novel therapies for endotheliopathy, hypercoagulability, and vasculitis are needed.
Conclusion
The endotheliopathy due to direct endothelial infection with SARS-COV-2 and the indirect damage caused by inflammation play the predominant role in the development of CAC. The intensive control of thromboinflammation is necessary to improve the outcome of this highly detrimental contagious disease.
Introduction
Ongoing reports have described the hypercoagulability and thrombotic tendency in COVID-19 [1]. The high incidence of deep vein thrombosis and pulmonary embolism has focused on the critical role of routine antithrombotic prophylaxis for COVID-19 management, especially in critically ill patients and/or elevated D-dimer levels [2,3,4]. Current reports of venous and arterial thrombotic events in the patients treated in ICU is up to 30% even with pharmacological thromboprophylaxis, and thrombotic events are associated with 5.4 times higher risk of mortality [5]. Recent postmortem evaluation of COVID-19 patients has demonstrated severe endothelial injury with cellular death/apoptosis, and the presence of intracellular virus in the autopsy lung with thrombosis and small to middle-size pulmonary vessels. The clotting and vascular damage were also confirmed in the alveolar capillary and these changes are more remarkable in COVID-19 compared to influenza induced lung injury [6]. In this summary, we will review the pathophysiology of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced endotheliopathy, COVID-19-associated coagulopathy (CAC), and vasculitis.
Coagulopathy in COVID-19
The mechanism of coagulopathy in COVID-19
Thromboembolic complications are the hallmark of COVID-19 that can cause death even in asymptomatic COVID-19 [7]. The new coronavirus SARS-CoV-2 elicits an acute inflammatory effect with hypercoagulability, platelet activation, and endothelial dysfunction [8]. Although, this presentation has similarities with sepsis-induced coagulopathy (SIC) due to bacterial infections and disseminated intravascular coagulation (DIC), there are several important differences [8]. In CAC, patients often initially present with increased fibrinogen levels, increased D-dimers, but minor changes in prothrombin time and platelet count compared to acute bacterial sepsis that can produce thrombocytopenia, prolonged prothrombin times, and decreased antithrombin levels [4, 9]. It is also known that inflammatory cytokine levels are elevated in COVID-19 and excess production of inflammatory cytokines can induce hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS) that can result in a thrombotic coagulation disorder [10]. Although the pathophysiology of HLH/MAS seems similar to COVID-19, the reported cytokine level is much lower in COVID-19 [11]. Conversely, the endothelial derangement, detailed in the next section, is predominant in COVID-19. Other than HLH/MAS, various thrombotic diseases such as thrombotic microangiopathy, and antiphospholipid syndrome can occur, and the characteristics of these diseases look similar to CAC [12]. Even though the pathogeneses of these thrombotic diseases partially overlap with CAC, it is important to delineate the unique statue of CAC to plan a therapeutic strategy.
The evaluation of COVID-19-associated coagulopathy
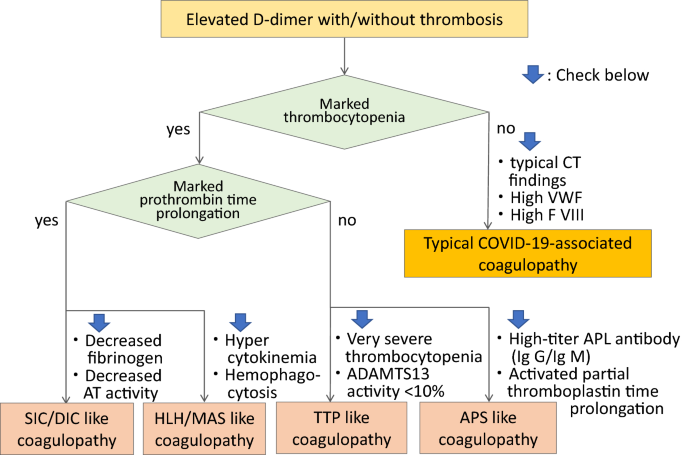
D-Dimer monitoring is important in COVID-19 coagulopathy. Although D-dimer is initially elevated, other conventional coagulation laboratory tests including prothrombin time (PT), activated partial thromboplastin time (aPTT), and platelet count are often normal, and are not useful indicators of the thrombotic risk. The increase of factor VIII and von Willebrand factor (VWF) [13], potentially the presence of antiphospholipid antibodies [14], and increased activity of complement system are also reported, however, monitoring these biomarkers is not practical. The pathogenesis of coagulopathy in COVID-19 is complex but the typical CAC can be diagnosed by increased D-dimer, elevated fibrinogen and VWF levels, but relatively normal PT, aPTT, and platelet count. A pathway for diagnosing CAC versus other coagulopathies is illustrated in Fig. 1.
Thrombin generation testing (TGT) measures ex vivo thrombin formation in plasma upon activation with tissue factor. TGT allows calculation of peak and total thrombin generation, as well as time to initial and peak thrombin generation. This assay can identify both reduced and increased thrombin generation. Nougier et al. [15] revealed increased thrombin generation in COVID-19 patients despite undergone anticoagulation. The major drawbacks of TGT are the lack of standardization and the requirement of technical training.
Other potential assays to assess global coagulation status include viscoelastic testing, especially in ICU patients, as a point of care test. An increasing number of studies report hypercoagulability as indicated by decreased R or clot times, and increased maximal amplitude/maximal clot firmness by viscoelastic monitoring [16,17,18]. However, these changes are consistent with high fibrinogen levels that affect both maximal amplitude on thromboelastography (TEG), and maximum clot firmness on rotational thromboelastometry (ROTEM). Ranucci et al. [17] reported the median fibrinogen level was nearly 800 mg/dL in the COVID-19 patients treated in ICU, and such a high fibrinogen level affects TEG and ROTEM parameters considerably by itself [19].
Endotheliopathy in COVID-19
The endothelial damage and thrombosis
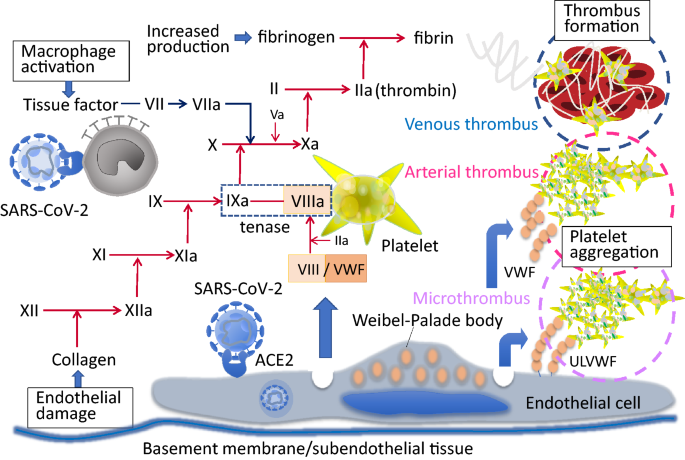
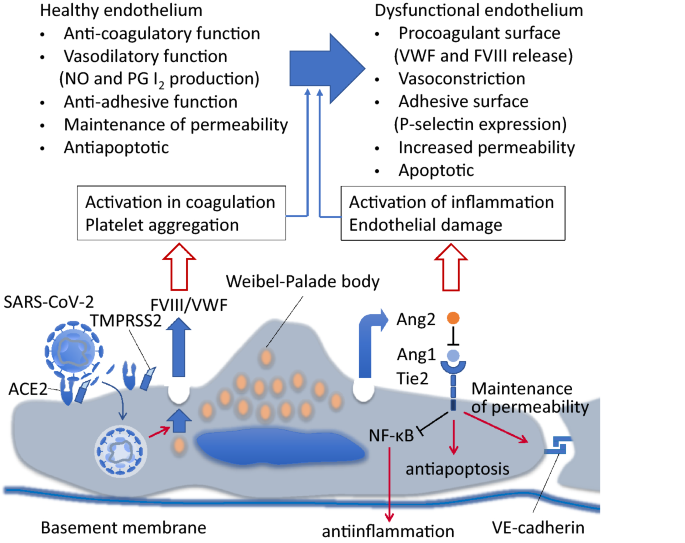
An important feature of CAC is the microcirculatory endothelial damage in pulmonary circulation and other vascular beds. Since SARS-CoV-2 directly infects the vascular endothelial cell causing cellular damage and apoptosis, the antithrombotic activity of the luminal surface is remarkably decreased [20]. In COVID-19, both alveolar damage and microcirculatory disturbance associated with thrombus formation contribute to respiratory dysfunction. At autopsy, findings reported include clot formation in pulmonary arterioles with diffuse alveolar damage and hyaline membranes [21]. Normal endothelial function refers to the ability of regulating vascular tonus, permeability, cell adhesion, and anticoagulation. Healthy endothelial cells synthesize nitric oxide (NO) by conversion of L-arginine to L-citrulline by nitric oxide synthase. NO released by endothelium prevents leukocyte and platelet adhesion, inflammatory cell migration into the vessel wall, smooth muscle cell proliferation, and suppresses apoptosis and inflammation. SARS-Cov-2 enters endothelial cells through endocytosis and is mediated by an interplay of Angiotensin-converting enzyme 2 (ACE2) and the transmembrane protease serine 2 (TMPRSS-2) which sheds a part of spike protein and helps SARS-Cov-2 to enter into endothelial cell. The infected endothelial cells lose their ability to maintain aforementioned physiological functions. Subsequently, the damage of the endothelium leads to the procoagulant change of the vascular lumen, formation of immunothrombosis, and organ malcirculation.
Both systemic pulmonary microthrombosis and thromboembolism are commonly seen in COVID-19. This typical figure is thought to be the result of hypercoagulability due to the dysregulated endothelial function of the pulmonary vessels and systemic inflammation. In addition to the deep vein thrombus that results in an embolic event, in situ formation in the pulmonary arteries can be the main reason of pulmonary dysfunction. Lax et al. [22] performed autopsies in 11 patients and reported that despite the absence of clinical presentations of thromboembolism, thrombus formation in small and mid-sized pulmonary arteries was found in all of the examined cases. In these cases, thrombus is suspected to form at the peripheral arteriole and elongate proximally. According to another series of autopsy findings, the incidence of thrombus formation in the pulmonary microvasculature is approximately nine times higher than that seen in influenza [6].
The endothelial damage-derived hypercoagulability
ACE2, the host cellular receptor of SARS-CoV-2, has been identified on the vascular endothelial surface. SARS-CoV-2 uses ACE2 to invade into the cell through the fusion of its membrane to the host cell membrane. As a result, the host cell loses ACE2 activity which subsequently leads to reduced angiotensin II inactivation and decreased conversion to antiotensin1-7. Increased angiotensin II stimulates vascular constriction and decreased antiotensin1-7 suppresses nitric oxide production which triggers increased thrombogenicity due to leucocyte and platelet adhesion and vasoconstriction [23].
The vascular endothelium is coated by a gel-like component known as the glycocalyx, that regulates vascular blood flow by providing an antithrombotic surface via antithrombin binding to the heparan sulfate constituents, a major component of the glycocalyx. Although the circulating antithrombin level has been reported to be in a normal range on presentation in COVID-19 cases [13], if the glycocalyx is disrupted, the local antithrombogenicity of the endothelial surface may be altered. However, little information on the glycocalyx status in COVID-19 is available.
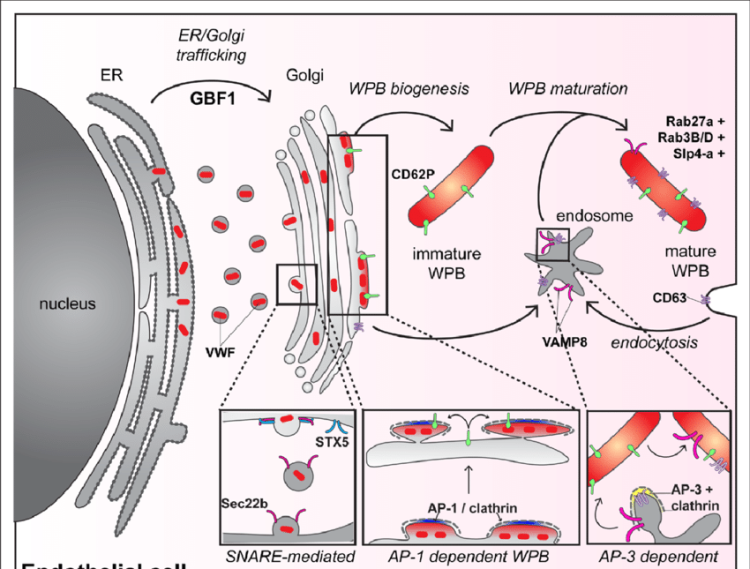
One of the unique features of CAC is the increase in VWF and factor VIII [13, 24] and it is suggested to be the result of vascular response to SARS-CoV-2 infection. VWF and factor VIII are stored in the Weibel-Palade body of endothelial cells and released in response to infectious stimuli [25] (Fig. 2). The increase in VWF suggests a possible similarity to thrombotic thrombocytopenic purpura, however, ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) levels in COVID-19 although reported to be decreased, may not be severely depleted as in thrombotic thrombocytopenic purpura [26]. The increased VWF levels to 3–4 times normal values seen in patients with COVID-19 may overwhelm the ADAMTS13 activity to degrade the ultra large VWF multimers. The importance of circulating VWF and multimer size in CAC is not established, however, one of the suggested methods to reduce the risk of thrombotic events due to excess ultra large VWF itself is plasma exchange [27, 28].
Similar to factor VIII and VWF, angiopoietin 2, also stored in Weibel-Palade bodies, is known to be released and its circulating level increases in COVID-19 [29]. Angiopoietin 2 serves as an antagonist of angiopoietin 1 and inhibits anti-inflammatory, anticoagulatory, and antiapoptotic signaling induced by angiopoietin 1 by binding to Tie2 competitively [30] (Fig. 3). Tie2 activation by angiopoietin 1 also normalizes prothrombotic responses by inhibiting endothelial tissue factor and phosphatidylserine exposure in sepsis, and therefore, Tie2 signaling is considered to play a central role in the regulation of thrombus formation in SIC/DIC [31]. Angiotensin 2 is also known to increase endothelial permeability and is considered an important factor in acute respiratory distress syndrome [32]. The role of angiopoietin 2 in CAC has not been studied well and it should be the focus in future studies.
The monitoring of endothelial damage
One of the difficulties in clinical studies of endothelial research is the limited availability of ideal biomarkers. The glycocalyx provides an interface between blood flow and endothelial cells. Since the glycocalyx is fragile, its components are used as biomarkers of endothelial damage in various diseases including infectious diseases [30]. One such biomarker is a hyaluronic acid, a major glycocalyx component. Circulating levels of hyaluronic acid, are elevated in critically ill COVID-19 patients compared with less severe cases [33]. Other than that, the proteins that are released from the Weibel-Palade body i.e., VWF, FVIII, and P-selectin are the potential biomarkers. In addition, sensitive coagulation markers such as thrombin-antithrombin complex (TAT) and prothrombin fragment 1 + 2 can be a marker for the microthrombosis. Goshua et al. [34] reported VWF antigen/antibody, FVIII activity, and TAT levels are significantly higher in the more severe cases.
As previously mentioned, angiopoietin 2 is stored in the endothelial cells and secreted along with the endothelial damage. Reportedly, angiopoietin 2 levels are associated with coagulation disorder, organ damage and death in bacterial sepsis [35]. Smadja et al. [29] measured angiopoietin 2, D-dimer, CRP, and creatinine in consecutive 40 COVID-19 patients treated in ICU, and found angiopoietin 2, cut-off of 5000 pg/mL, as the best predictor for poor outcome (sensitivity: 80.1%, specificity: 70%). It is crucial to find a good biomarker of vascular damage in COVID-19 study.
Therapeutic strategies for endothelial damage
Despite prophylactic anticoagulation in CAC, patients can still develop thrombotic sequela. A recent study reported that despite systematic use of thromboprophylaxis, 31% of the COVID-19 patients treated in ICU developed thrombotic complications [36]. In another study, the cumulative incidence of arterial and venous thromboembolism was 49% [3]. These reports suggest that despite anticoagulation, additional therapy for endothelial injury is necessary to prevent thrombosis. Potential therapies include synthetic serine protease inhibitors such as nafamostat mesylate and camostat mesylate which theoretically prevent SARS-CoV-2 infection. Coronavirus gains entry to the cell using the host TMPRSS2 which cleaves the spike protein resulting in its ability to fuse to the host cellular membrane. These agents inhibit TMPRSS2 thereby abrogating the activating proteolytic processing of virus [37]. Since nafamostat mesylate also has anticoagulatory effects, it has been used for DIC and anticoagulation for extracorporeal circuits in Japan.
Other therapeutic considerations are the physiologic anticoagulants such as protein C and antithrombin. The dual action of protein C/activated protein C to inactivate factor VIIIa and upregulate ACE2 are the advantage of this system which suppress both coagulation and inflammation. Activated protein C can also reduce pulmonary injury by suppressing the macrophage inflammatory protein family chemokine response [38]. Antithrombin is another multifaceted serine protease inhibitor of multiple coagulation factors, but also protects the glycocalyx by binding to heparan sulfate [39]. Bikdeli et al. [40] noted in their recent review of pharmacological therapy targeting thromboinflammation in COVID-19, that antithrombin suppresses excess inflammation by inhibiting nuclear factor-κB, it may be suitable for the treatment of CAC. However, the effects of these agents in COVID-19 haven’t been examined in clinical trials and future study may be warranted.
Arterial thrombosis in COVID-19
Arterial thrombosis is an uncommon event in other infection-associated coagulopathies. In contrast, stroke, ischemic coronary disease, and thrombotic limb ischemia can occur in COVID-19. Lodigiani et al. [41] reported the rate of ischemic stroke and acute coronary syndrome was 2.5% and 1.1%, respectively, in Italy. Kashi et al. [42] reported two cases of floating thrombi in thoracic aorta and such cases are extremely odd in previously described infectious diseases. Antiphospholipid syndrome is known as a disease that result in arterial thrombosis and can occur secondary to infection. Some reports have shown increased lupus anticoagulant, anticardiolipin, and anti-β2-glycoprotein I antibodies, however, the presence of high titer IgG antibody, an important responsible factor, has not proven yet [13, 24, 43], and the association between antiphospholipid syndrome and CAC is still unclear. The presence of unusually large VWF multimers and subsequent activation of platelets and microthrombi can explain the occurrence of arterial macrothrombosis [44], and Williams et al. [45] reported elevated VWF levels were associated with the increased risk for recurrent stroke. However, a definitive cause and effect relation has not been proven yet. The occurrence of arterial thrombosis is difficult to predict and there are no good prophylactic strategies. Oxley et al. [7] reported five cases of large-vessel stroke in patients younger than 50 years of age. The mechanism of arterial thrombosis in COVID-19 remains a mystery and prediction of the events was not possible in any of the cases; demographic factors, laboratory data, and severity of COVID-19 did not appear to be related to arterial events.
Clot formation in extracorporeal circuits
The high incidence of clot formation during extracorporeal circulation has been recognized. Helms et al. [13] studied 150 COVID-19 patients and reported 28 out of 29 patients (96.6%) receiving continuous renal replacement therapy (CRRT) experienced clotting of the circuit. The median lifespan of an CRRT circuit was 1.5 days which is only half of the recommendation duration. They also reported 12 patients (8%) were treated by extracorporeal membrane oxygenation (ECMO), and among them, thrombotic occlusions of centrifugal pump occurred in 2 patients. Methods to minimize extracorporeal circuit clotting include prefilter infusion of heparin and the use of citrate-based replacement fluid for dialysis are not always successful [46]. The reason for the high incidence of CRRT filter and ECMO oxygenator coagulation is not known, but factors other than endotheliopathy such as hypercoagulability, hypofibrinolysis, and platelet activation must attribute. The elevated VWF activity, increased factor VIII level, and high fibrinogen level may lead to microthrombi formation possibly occluding the filter. Suppressed fibrinolysis may also play a role [47], with excess angiotensin II enhancing the expression of PAI-1 in the endothelium in COVID-19 [48].
Vasculitis in COVID-19
A report from northern Italy observed significantly increased number of patients with Kawasaki disease, an acute self-limiting vasculitis predominantly involving the coronary arteries, with hemodynamically unstable Kawasaki disease shock syndrome (KDSS) during the COVID-19 pandemic [49]. It is also reported that children with COVID-19 are more likely to show MAS that resembles secondary HLH. Varga et al. [50] demonstrated the direct viral infection of the endothelial cell and diffuse endothelial inflammation which are followed by the induction of endothelitis, apoptosis, and pyroptosis in autopsy cases of COVID-19. Of note is the mononuclear cell infiltrations into the vascular intima along the lumen of many vessels also reported in this post-mortem analysis. This finding suggests that the virus can invade into human vasculature and cause vasculitis. Roncati et al. [51] estimated the escalation from type 2 T-helper immune response to type 3 hypersensitivity is involved in the pathophysiology of COVID-19-induced vasculitis. They reported the deposition of immune complexes inside the vascular walls causing more severe inflammatory reaction, and interleukin-6 is the key myokine in this scenario.
Conclusion
The mechanism of coagulopathy in COVID-19 continues to be investigated. However, the predominant role of endotheliopathy due to direct endothelial infection with SARS-COV-2 and the indirect damage caused by inflammation are part of the complex thromboinflammatory process. The elevated circulating levels of clotting factors including fibrinogen, factor VIII, VWF released from the stimulated endothelial cells, and the loss of the thromboprotective function with glycocalyx damage and decreased nitric oxide production also contribute to the coagulopathy and thromboinflammation. SARS-CoV-2 damages not only the luminal surface of the vasculature but also induce vasculitis, contributing to the significant pathology associated with COVID-19.
References
- 1.Han H, Yang L, Liu R. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med. 2020. https://doi.org/10.1515/cclm-2020-0188.Article PubMed Google Scholar
- 2.Ren B, Yan F, Deng Z, Zhang S, Xiao L, Wu M, Cai L. Extremely high incidence of lower extremity deep venous thrombosis in 48 patients with severe COVID-19 in Wuhan. Circulation. 2020. https://doi.org/10.1161/CIRCULATIONAHA.120.047407.Article PubMed Google Scholar
- 3.Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, Clark C, Iba T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020;18(5):1023–6.CAS Article Google Scholar
- 4.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020:blood.2020006000.
- 5.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb Res. 2020;S0049–3848(20):30157–62.Google Scholar
- 6.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2015432.Article PubMed Google Scholar
- 7.Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, Skliut M, Weinberger J, Dangayach NS, Bederson JB, Tuhrim S, Fifi JT. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382(20):e60.Article Google Scholar
- 8.Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7(6):e438–e440440.Article Google Scholar
- 9.Iba T, Levy JH. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Anesthesiology. 2020;132(5):1238–45.Article Google Scholar
- 10.McGonagle D, et al. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020. https://doi.org/10.1016/S2665-9913(20)30121-1.Article PubMed PubMed Central Google Scholar
- 11.Leisman DE, Deutschman CS, Legrand M. Facing COVID-19 in the ICU: vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. 2020;28:1–4. https://doi.org/10.1007/s00134-020-06059-6.CAS Article Google Scholar
- 12.Opoka-Winiarska V, Grywalska E, Roliński J. Could hemophagocytic lymphohistiocytosis be the core issue of severe COVID-19 cases? BMC Med. 2020;18(1):214.CAS Article Google Scholar
- 13.Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Fagot Gandet F, Fafi-Kremer S, Castelain V, Schneider F, Grunebaum L, Anglés-Cano E, Sattler L, Mertes PM, Meziani F, CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis). High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020:1–10.
- 14.Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, Wang C, Zhao J, Sun X, Tian R, Wu W, Wu D, Ma J, Chen Y, Zhang D, Xie J, Yan X, Zhou X, Liu Z, Wang J, Du B, Qin Y, Gao P, Qin X, Xu Y, Zhang W, Li T, Zhang F, Zhao Y, Li Y, Zhang S. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med. 2020;382(17):e38.Article Google Scholar
- 15.Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, David JS, Bonnet A, Negrier C, Dargaud Y. Hypofibrinolytic state and high thrombin generation may play a major role in sars-cov2 associated thrombosis. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.15016.Article PubMed PubMed Central Google Scholar
- 16.Maatman TK, Jalali F, Feizpour C, Douglas A 2nd, McGuire SP, Kinnaman G, Hartwell JL, Maatman BT, Kreutz RP, Kapoor R, Rahman O, Zyromski NJ, Meagher AD. Routine venous thromboembolism prophylaxis may be inadequate in the hypercoagulable state of severe coronavirus disease 2019. Crit Care Med. 2020. https://doi.org/10.1097/CCM.0000000000004466.Article PubMed PubMed Central Google Scholar
- 17.Ranucci M, Ballotta A, Di Dedda U, Bayshnikova E, Dei Poli M, Resta M, Falco M, Albano G, Menicanti L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14854.Article PubMed PubMed Central Google Scholar
- 18.Panigada M, Bottino N, Tagliabue P, Grasselli G, Novembrino C, Chantarangkul V, Pesenti A, Peyvandi F, Tripodi A. Hypercoagulability of COVID-19 patients in Intensive Care Unit. A report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14850.Article PubMed Google Scholar
- 19.Scala E, Coutaz C, Gomez F, Alberio L, Marcucci C. Comparison of ROTEM sigma to standard laboratory tests and development of an algorithm for the management of coagulopathic bleeding in a tertiary center. J Cardiothorac Vasc Anesth. 2020;34(3):640–9.CAS Article Google Scholar
- 20.Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Schröder AS, Burdelski C, de Heer G, Nierhaus A, Frings D, Pfefferle S, Becker H, Bredereke-Wiedling H, de Weerth A, Paschen HR, Sheikhzadeh-Eggers S, Stang A, Schmiedel S, Bokemeyer C, Addo MM, Aepfelbacher M, Püschel K, Kluge S. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. 2020;6:M20–2003. https://doi.org/10.7326/M20-2003.Article Google Scholar
- 21.Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, da Silva LFF, de Oliveira EP, Nascimento Saldiva PH, Mauad T, Marcia NE. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14844.Article PubMed PubMed Central Google Scholar
- 22.Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, Vander K, Bargfrieder U, Trauner M. Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med. 2020. https://doi.org/10.7326/M20-2566.Article PubMed PubMed Central Google Scholar
- 23.Verdecchia P, Cavallini C, Spanevello A, Angeli F. COVID-19: ACE2 centric infective disease? Hypertension. 2020. https://doi.org/10.1161/HYPERTENSIONAHA.120.15353.Article PubMed Google Scholar
- 24.Escher R, Breakey N, Lämmle B. ADAMTS13 activity, von Willebrand factor, factor VIII and D-dimers in COVID-19 inpatients. Thromb Res. 2020;192:174–5.CAS Article Google Scholar
- 25.Streetley J, Fonseca AV, Turner J, Kiskin NI, Knipe L, Rosenthal PB, Carter T. Stimulated release of intraluminal vesicles from Weibel–Palade bodies. Blood. 2019;133(25):2707–17.CAS Article Google Scholar
- 26.Huisman A, Beun R, Sikma M, Westerink J, Kusadasi N. Involvement of ADAMTS13 and von Willebrand factor in thromboembolic events in patients infected with SARS-CoV-2. Int J Lab Hematol. 2020. https://doi.org/10.1111/ijlh.13244.Article PubMed PubMed Central Google Scholar
- 27.Keith P, Day M, Perkins L, Moyer L, Hewitt K, Wells A. A novel treatment approach to the novel coronavirus: an argument for the use of therapeutic plasma exchange for fulminant COVID-19. Version 2. Crit Care. 2020;24(1):128.Article Google Scholar
- 28.Zachariah U, Nair SC, Goel A, Balasubramanian KA, Mackie I, Elias E, Eapen CE. Targeting raised von Willebrand factor levels and macrophage activation in severe COVID-19: consider low volume plasma exchange and low dose steroid. Thromb Res. 2020;192:2.CAS Article Google Scholar
- 29.Smadja DM, Guerin CL, Chocron R, Yatim N, Boussier J, Gendron N, Khider L, Hadjadj J, Goudot G, Debuc B, Juvin P, Hauw-Berlemont C, Augy JL, Peron N, Messas E, Planquette B, Sanchez O, Charbit B, Gaussem P, Duffy D, Terrier B, Mirault T, Diehl JL. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis. 2020;27:1–10. https://doi.org/10.1007/s10456-020-09730-0.CAS Article Google Scholar
- 30.Uchimido R, Schmidt EP, Shapiro NI. The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit Care. 2019;23(1):16.Article Google Scholar
- 31.Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, Schulman S, Libermann TA, Lu S, Shapiro NI, Christiani DC, Flaumenhaft R, Parikh SM. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest. 2018;128(4):1471–84.Article Google Scholar
- 32.Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. Version 2. PLoS Med. 2006;3(3):e46.Article Google Scholar
- 33.Ding M, Zhang Q, Li Q, Wu T, Huang YZ. Correlation analysis of the severity and clinical prognosis of 32 cases of patients with COVID-19. Respir Med. 2020;167:105981.Article Google Scholar
- 34.Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, Dela Cruz CS, Dumont A, Halene S, Hwa J, Koff J, Menninger H, Neparidze N, Price C, Siner JM, Tormey C, Rinder HM, Chun HJ, Lee AI. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7(8):e575–e582582.Article Google Scholar
- 35.Fisher J, Douglas JJ, Linder A, Boyd JH, Walley KR, Russell JA. Elevated plasma angiopoietin-2 levels are associated with fluid overload, organ dysfunction, and mortality in human septic shock. Crit Care Med. 2016;44(11):2018–27.CAS Article Google Scholar
- 36.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020:S0049-3848(20)30120-1.
- 37.Yamaya M, Nishimura H, Deng X, Kikuchi A, Nagatomi R. Protease inhibitors: candidate drugs to inhibit severe acute respiratory syndrome coronavirus 2 replication. Tohoku J Exp Med. 2020;251(1):27–30.CAS Article Google Scholar
- 38.Richardson MA, Gupta A, O’Brien LA, Berg DT, Gerlitz B, Syed S, Sharma GR, Cramer MS, Heuer JG, Galbreath EJ, Grinnell BW. Treatment of sepsis-induced acquired protein C deficiency reverses Angiotensin-converting enzyme-2 inhibition and decreases pulmonary inflammatory response. J Pharmacol Exp Ther. 2008;325(1):17–26.CAS Article Google Scholar
- 39.Iba T, Levy JH, Hirota T, et al. Protection of the endothelial glycocalyx by antithrombin in an endotoxin-induced rat model of sepsis. Thromb Res. 2018;171:1–6.CAS Article Google Scholar
- 40.Bikdeli B, Madhavan MV, Gupta A, Jimenez D, Burton JR, Der Nigoghossian C, Chuich T, Nouri SN, Dreyfus I, Driggin E, Sethi S, Sehgal K, Chatterjee S, Ageno W, Madjid M, Guo Y, Tang LV, Hu Y, Bertoletti L, Giri J, Cushman M, Quéré I, Dimakakos EP, Gibson CM, Lippi G, Favaloro EJ, Fareed J, Tafur AJ, Francese DP, Batra J, Falanga A, Clerkin KJ, Uriel N, Kirtane A, McLintock C, Hunt BJ, Spyropoulos AC, Barnes GD, Eikelboom JW, Weinberg I, Schulman S, Carrier M, Piazza G, Beckman JA, Leon MB, Stone GW, Rosenkranz S, Goldhaber SZ, Parikh SA, Monreal M, Krumholz HM, Konstantinides SV, Weitz JI, Lip GYH, Global COVID-19 Thrombosis Collaborative Group. Pharmacological agents targeting thromboinflammation in COVID-19: review and implications for future research. Thromb Haemost. 2020. https://doi.org/10.1055/s-0040-1713152.Article PubMed PubMed Central Google Scholar
- 41.Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD, Sacco C, Alexia B, Sandri MT, Barco S, Humanitas COVID-19 Task Force. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020;191:9–14.CAS Article Google Scholar
- 42.Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res. 2020;190:62.CAS Article Google Scholar
- 43.Connell NT, Battinelli EM, Connors JM. Coagulopathy of COVID-19 and antiphospholipid antibodies. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14893.Article PubMed PubMed Central Google Scholar
- 44.Chang JC. Acute respiratory distress syndrome as an organ phenotype of vascular microthrombotic disease: based on hemostatic theory and endothelial molecular pathogenesis. Clin Appl Thromb Hemost. 2019;25:1076029619887437.CAS Article Google Scholar
- 45.Williams SR, Hsu FC, Keene KL, Chen WM, Dzhivhuho G, Rowles JL 3rd, Southerland AM, Furie KL, Rich SS, Worrall BB, Sale MM. Genetic drivers of von Willebrand factor levels in an ischemic stroke population and association with risk for recurrent stroke. Stroke. 2017;48(6):1444–500.CAS Article Google Scholar
- 46.Sise ME, Baggett MV, Shepard JO, Stevens JS, Rhee EP. Case 17–2020: a 68-year-old man with Covid-19 and acute kidney injury. N Engl J Med. 2020;382(22):2147–56.Article Google Scholar
- 47.Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S, Nydam TL, Moore PK, McIntyre RC Jr. Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. J Am Coll Surg. 2020;S1072–7515(20):30400–2.Google Scholar
- 48.Kwaan HC. Coronavirus disease 2019: the role of the fibrinolytic system from transmission to organ injury and sequelae. Semin Thromb Hemost. 2020. https://doi.org/10.1055/s-0040-1709996.Article PubMed PubMed Central Google Scholar
- 49.Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, Bonanomi E, D’Antiga L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020. https://doi.org/10.1016/S0140-6736(20)31103-X.Article PubMed PubMed Central Google Scholar
- 50.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8.CAS Article Google Scholar
- 51.Roncati L, Ligabue G, Fabbiani L, Malagoli C, Gallo G, Lusenti B, Nasillo V, Manenti A, Maiorana A. Type 3 hypersensitivity in COVID-19 vasculitis. Clin Immunol. 2020;29:108487. https://doi.org/10.1016/j.clim.2020.108487.CAS Article Google Scholar
Funding
This work was supported in part by a Grant-in-Aid for Special Research in Subsidies for ordinary expenses of private schools from The Promotion and Mutual Aid Corporation for Private Schools of Japan.
Author information
Affiliations
- Department of Emergency and Disaster Medicine, Juntendo University Graduate School of Medicine, 2-1-1 Hongo Bunkyo-ku, Tokyo, 113-8421, JapanToshiaki Iba
- Hematology Division Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USAJean Marie Connors
- Department of Anesthesiology, Critical Care, and Surgery, Duke University School of Medicine, Durham, NC, USAJerrold H. Levy