Authors: Gabriela Athziri Sánchez-Zuno,1,†Mónica Guadalupe Matuz-Flores,1,†Guillermo González-Estevez,1Ferdinando Nicoletti,2Francisco Javier Turrubiates-Hernández,1Katia Mangano,2 and José Francisco Muñoz-Valle1
Int J Immunopathol Pharmacol. 2021 Jan-Dec; 35: 20587384211050199.Published online 2021 Oct 10. doi: 10.1177/20587384211050199PMCID: PMC8512237PMID: 34632844
Abstract
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), represents an unprecedented global public health emergency with economic and social consequences. One of the main concerns in the development of vaccines is the antibody-dependent enhancement phenomenon, better known as ADE. In this review, we provide an overview of SARS-CoV-2 infection as well as the immune response generated by the host. On the bases of this principle, we also describe what is known about the ADE phenomenon in various viral infections and its possible role as a limiting factor in the development of new vaccines and therapeutic strategies.
Keywords: COVID-19, SARS-CoV-2, ADE, vaccine, antibody-dependent enhancement
Introduction
The first cases of coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), were identified in December 2019 in China. The spread of this disease occurred rapidly throughout the world mainly due to its forms of transmission, being the most important the contact with respiratory fluids (exposure to respiratory droplets carrying infectious viruses). 1
Following the outbreak in China, a trend of increasing cases grew exponentially as it was observed. As a consequence of this rapid spread, the World Health Organization (WHO) declared COVID-19 as an international public health emergency, and in March 2020, it was declared a pandemic. As a result of this unexpected progression, health authorities around the world entered into a state of alert facing the need to implement unprecedented sanitation and isolation protocols. 2
The state of pandemic has caused a great impact on both the economic and public health level around the world, because of social distancing, border closures, and the performance of essential activities only. According to the WHO, in the week of April 12th to April 20th, 2021, more than 140 million cases and more than 3 million deaths have already been reported worldwide, new cases continued to rise globally, in the past week to over 5.2 million new reported cases. Possible reasons for this increase include the continued spread of more transmissible variants of concern (VOCs). The countries such as India, the United States of America, Brazil, Turkey, and France reported the highest number of cases. 2
Regarding the health implications of this disease, it has been described that the majority of patients infected by COVID-19 have symptoms of a common cold such as fever, cough, fatigue, headache, and muscle pain as well as diarrhea. In some cases, severe shortness of breath can also occur. 3 Although most patients have a favorable prognosis, in some cases this may not be the scenario. A poor prognosis has been associated with the presence of some chronic diseases and comorbidities including hypertension, diabetes, coronary heart disease, and obesity. In the case of diabetes, patients are more susceptible to developing the so-called “cytokine storm” that leads to a rapid deterioration of COVID-19.4,5
Another important aspect regarding the pathogenesis of COVID-19 is the occurrence of the phenomenon called antibody-dependent enhancement (ADE). This mechanism involves endocytosis of virus–antibody immune complexes into cells through interaction of the antibody Fc region with cellular Fc receptors (FcRs). In this event, pre-existing non-neutralizing or sub-neutralizing antibodies to viral surface proteins that were generated during a previous infection can promote the subsequent entry of viruses into the cell and therefore intensify the inflammatory process during a secondary infection with any antigenic-related virus.6–8 The occurrence of ADE may represent one of the greatest challenges for scientists working on the development of a safe vaccine against COVID-19.
For the aforementioned, in this review, we provide an overview of SARS-CoV-2 infection as well as the immune response generated by the host. On the bases of this principle, we also describe what is known about the ADE phenomenon in various viral infections and its role as a limiting factor in the development of new vaccines and therapeutic strategies.
Structure and pathogenesis of SARS-CoV-2
The Coronaviruses (CoVs) are viruses that show morphological similarity to a solar corona appearance under an electron microscope due to the presence of “spike” glycoproteins. These CoVs belong to the large family Coronaviridae, which consists of two subfamilies: Orthocoronavirinae and Torovirinae. The Orthocoronavirinae subfamily is classified into four genera: alpha coronaviruses, beta coronaviruses, gamma coronaviruses, and delta coronaviruses. Among these, the beta genus is the one that has been described as capable of causing severe illness and even death among infected individuals.9,10
The genome of this beta-CoV has been classified as a single-stranded ribonucleic acid (RNA) virus consisting of 26–32 kbp and contains 7–10 open reading frames (ORF). Two-thirds of the genome encodes the replicase-transcriptase proteins, and a third part encodes the four structural proteins: spike (S), envelope, membrane, and nucleoprotein. The S-glycoproteins on the surface of CoVs comprise the receptor-binding domain(s) and contribute for host cell binding, host–viral cell membrane fusion, and virus internalization while the M-glycoprotein plays a role in the virion envelope formation and assembly.9–12 Therefore, the entry of the coronavirus into susceptible cells is a complex process that requires receptor binding and proteolytic processing of protein S to promote virus–cell fusion. As anticipated above, SARS-CoV-2 is acquired by exposure to respiratory fluids of infected individuals and less through contact with fomites. 13
SARS CoV interacts directly with angiotensin-converting enzyme 2 (ACE2) to enter target cells. At the onset of the infection, SARS-CoV-2 targets mainly host cells that express ACE2, including bronchial cells, airway epithelial cells, alveolar epithelial cells, macrophages in the lung, and vascular endothelial cells. 14
After the recognition and binding of the SARS-CoV-2 S-glycoprotein with ACE2 in the host cells, the S-protein is cleaved by transmembrane protease serine 2 (TMPRSS2) to reveal the S2 domain necessary for the fusion of the viral membrane–host cell and the entry of the virus. Once the viral content is released into host cells, the viral RNA that enters, begins its replication, production, and release of new viral particles (Figure 1(a)).14,15

Open in a separate windowFigure 1.
{kind=link}
The Immune Response and Immunopathology of COVID-19. (a) The entry of SARS-CoV-2 into cells is mediated by the binding of TMPRSS2 and S-glycoprotein with the ACE2 acting as a receptor that facilitates viral binding to the membrane of the host cells. The virus enters by endocytosis and releases its RNA, replicates and creates new virions that cause a rapid progression of the infection. (b) Bronchial epithelial cells, type I and type II alveolar pneumocytes, and capillary endothelial cells become infected and a response occurs that leads to recruitment of macrophages, monocytes, neutrophils, and cytokine production in response to virus entry. (c) Sub-epithelial dendritic cells recognize the virus antigen and present them to CD4 + T cells that induce the differentiation of B cells into plasma cells that promote the production of virus-specific antibodies. Neutralizing antibodies can interact with phagocytes and NK cells and enhance antibody-mediated clearance of SARS-CoV. (d) A dysfunctional immune response leads to excessive cell infiltration, cytokine storm, inflammation, apoptosis, and multi-organ damage. Ab, antibody; ACE2, angiotensin-converting enzyme 2; FcγR, Fcγ receptor; IL, interleukin; MHC, major histocompatibility complex; TCR, T-cell receptor; TMPRSS2, transmembrane protease serine 2; TNF-a, tumor necrosis factor.
Innate immune response
After SARS-CoV-2 enters the host cells, it is recognized by pattern recognition receptors (PRRs) such as Toll-like receptor-7 (TLR7) and TLR8, which are expressed by epithelial cells that activate the local immune response, recruiting macrophages and monocytes that respond to infection (Figure 1(b)).
Once SARS-CoV-2 binds to PRRs, the recruited adapter proteins activate transcription factors. This includes interferon regulatory factor (IRF) and Nuclear factor κB (NF-κB), that lead to the production of antiviral type I interferon (IFN), and cytokines that induce an alarm signal in neighboring cells to attract other cells of innate immunity including polymorphonuclear cells, natural killer cells (NKs), dendritic cells, and monocytes.16,17 One of the signature features of this disease in patients with worst prognosis is the high serum levels of cytokines such as IL-1β, IL-6, TNF, IL1RA, and IL-8. These cytokines have an important role in the exacerbation of the inflammatory process and lead to the recruitment of other immune cells such as neutrophils and T cells. Among infiltrated innate cells, neutrophils can promote the destruction of viruses, but they can also worsen disease progression by inducing severe lung lesions.18,19
Types I and III IFNs are considered to be crucial in the antiviral response, and SARS-CoV-2 has been shown to be sensitive to pretreatment with IFN-I and III in vitro assays.20,21 The IFN timing and location are a key factor for an effective response against the virus. A study of the Middle East respiratory syndrome (MERS) in mice demonstrated that blockade of IFN signaling leads to a delayed virus clearance with increased neutrophil infiltration and alteration in T cell response. Conversely, 1 day of IFN-I administration protected mice from lethal infection, meanwhile, delayed IFN treatment failed to inhibit the replication of the virus. 22
One of the most important questions that arises in relation to innate immunity is how the SARS-CoV-2 evades the immune response. In a recent study, Kaneko et al., propose that the evasion of the antiviral aspects of innate immunity and the inflammatory process as a consequence of the virus can probably result in an alteration of the environment that leads to the attenuation of immunity of CD8 + T cells. In addition, there is an absence of germinal centers with reduction of B cells; therefore, it gives rise to a memory with a short duration and to B cells without high affinity. So far, it is still a very difficult question to answer. 23 However, it has been shown that patients with COVID-19 with worst prognosis showed poor IFN-I signals compared to patients with a favorable prognosis. 24
Additionally, various evasion mechanisms have been described for CoVs, with viral factors that antagonize pathways from PRR detection, cytokine secretion, and IFN signal induction. CoVs are able to evade PRRs by protecting the double strand RNA (dsRNA) with membrane-bound compartments formed during viral replication. Furthermore, SARS-CoV-2 is protected with guanosine and methylated by nonstructural proteins. They resemble host mRNA to promote translation, prevent degradation, and avoid detection of RIG-I-like receptors (RLRs).25–27
Adaptive immune response
The main mechanisms for decreasing viral replication, limit virus spread, and inflammation include the production of various pro-inflammatory cytokines, the activation of CD4 + and CD8 + T cells.28,29
The mechanism for the presentation of viral peptides occurs once the virus is inside respiratory cells. They are presented through the major histocompatibility complex (MHC) class I for cytotoxic CD8+ T cells which are essential to mediate elimination of cells infected by the virus. Additionally, the virus and its viral particles can be presented in the context of MHC class II by means of antigen-presenting cells, including dendritic cells and macrophages. They are in charge of presenting viral proteins to CD4+ T cells that provide the signals necessary for the induction of B cells and differentiation of plasma cells producing virus-specific neutralizing antibodies (Figure 1(c)).28,29
However, in patients with COVID-19, a low count of lymphocytes, CD4+ T cells, CD8+ T cells, B cells, and NK cells has been shown. Likewise, severe cases have presented lower levels of these cells compared to mild cases. 30 Secretion of type I IFNs dramatically increases the response of CD8 + T cells against viruses, but SARS-CoV-2 has been shown to possess nonstructural proteins that induce a decreased response to type I interferon (IFN) in infected cells. Therefore, the decrease in type I IFNs by different non-structural proteins of SARS-CoV-2 could explain the marked absence of CD8+ T cell response in COVID-19 patients.31–33
Kaneko et al., evaluated subsets of CD4+ T cells in lymph nodes and the spleen and observed that TH1 cells increase steadily at the beginning and end in lymph nodes and the spleen, also, a constant decrease in TH2 cells was described. Furthermore, FOXP3 + T reg cells make up a large part of the CD4+ T cell population at the end of disease. 23 Furthermore, it was shown that patients with significant decreases in T cell counts, especially CD8+ T cells, have elevated levels of IL-6, IL-10, IL-2, and IFN-γ in the peripheral blood. 34
Elevated cytokine secretion promotes cell infiltration inflammatory by establishing an aberrant inflammatory feedback loop that can cause damage to the lung. It can also cause damage through the secretion of proteases and reactive oxygen species (ROS) with subsequent alveolar damage and desquamation of alveolar cells. This results in inefficient gas exchange in the lung, which is reflected in low oxygen levels in patients. 35
Overall, impaired acquired immune responses and uncontrolled innate inflammatory responses to SARS-CoV-2 can cause cytokine storms that are associated with COVID-19 severity states and can lead to migration to different organs, causing multi-organ damage (Figure 1(d)). 36
Antibody responses in COVID-19 patients occur in conjunction with CD4+ T cell responses that induce B cells to differentiate into plasma cells and subsequently produce antibodies. In patients with SARS-CoV infection, the main target of neutralizing antibodies is the virus S glycoprotein, particularly with its receptor-binding domain (RBD), which is responsible for the binding of the virus to the ACE2 in host cells. 37 Neutralizing antibody responses to protein S possibly begin to develop in week two, and in most patients, antibody titers are detected by the third week.38,39
A recent study conducted by Ni et al., 2020 40 showed the presence of specific IgM and IgG antibodies for the structural proteins N (nuclear) and S-RBD in serum of recently negative COVID-19 patients compared to healthy donors. The IgG anti-SARS-CoV-2 was also higher in titers than IgM in follow-up patients compared to healthy donors. This indicates that patients with COVID-19 have IgG- and IgM-mediated responses to SARS-CoV-2 proteins, especially N and S-RBD. It also proposes that previously infected patients could maintain their IgG levels for at least 2 weeks after receiving a negative COVID-19 test result. 40Go to:
The devil in disguise: What happens when antibodies go bad
All viruses initiate infection by adhering to host cells through the interaction between viral proteins and receptor/coreceptor molecules on target cells (Figure 2(a)) As mentioned above, the host’s humoral response is responsible for generating specific antibodies to surface proteins that inhibit this step of the infection cycle, resulting in virus neutralization. Conversely, in some cases, these antibodies may paradoxically favor the infection process as part of a phenomenon better known as antibody-dependent enhancement (ADE). 41

{kind=link}
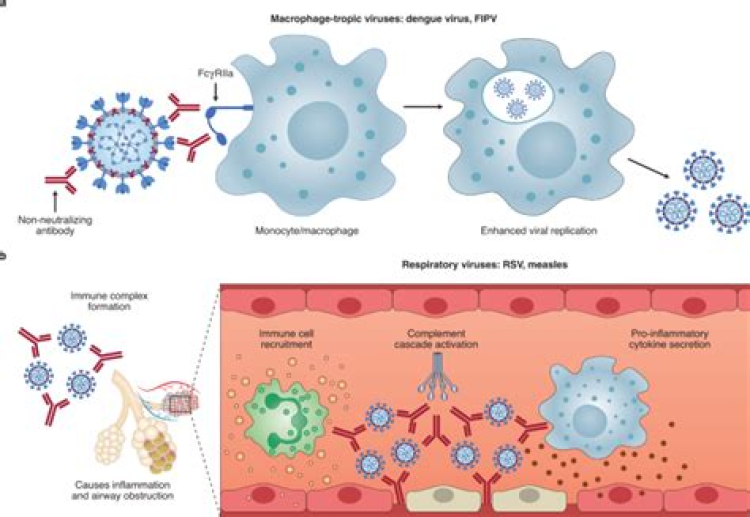
ADE phenomenon. (a) The conventional mechanism of infection by SARS-CoV 2 consists of the binding of its S-protein to the cellular receptor ACE2. After the union of the SARS-CoV-2 virus to the receptor, a conformational change occurs in the S-protein necessary for the fusion of the viral envelope with the cell membrane for subsequent endocytosis. Subsequently, SARS-CoV-2 releases its genetic material into the host cell. The RNA of the viral genome is then translated into proteins necessary for the subsequent assembly of viriomes in the ER and Golgi. These visions are then transported through vesicles outside the cell by exocytosis. The ADE phenomenon can be classified as two different mechanisms: ADE through enhanced infection and ADE through enhanced immune activation. (b) In ADE through increased infection, antibodies of a non-neutralizing or sub-neutralizing nature cause viral infection through FcγRIIa-mediated endocytosis, resulting in a more severe disease phenotype. (c) In ADE via enhanced immune activation, non-neutralizing antibodies can form immune complexes with viral antigens inside airway tissues, resulting in the secretion of pro-inflammatory cytokines, immune cell recruitment, and activation of the complement cascade within lung tissue. ADE, antibody-dependent enhancement; ACE2, angiotensin-converting enzyme 2; CR, compliment receptor; ER, endoplasmic reticulum; FcγRIIa, Fc γ receptor IIa; IFN-a, interferon a; IL, interleukin; IRF, interferon regulatory factors; iNOS, inducible nitric oxide synthase; PGE2, prostaglandin E2, RNA, ribonucleic acid; TNF-a, tumor necrosis factor.
Regarding the mechanism of ADE, it has been described that it involves endocytosis of virus–antibody immune complexes into cells through interaction of the antibody Fc region with cellular Fc receptors (FcRs). It is well known that the FcγRI (CD64) binds with high affinity to IgG monomerically while FcγRII (CD32) and FcγRIII (CD16) do so with low affinity and are activated by immune complexes. 42 In this regard, it is postulated that myeloid cells that express FcRs such as monocytes and macrophages, dendritic cells, and certain granulocytes can promote ADE through phagocytic uptake of the immune complexes. Although ADE is principally mediated by IgG antibodies, IgM along with complement, and IgA antibodies have also been described as capable of ADE 43
The phenomenon of ADE is an event that occurs in some viruses, where pre-existing non-neutralizing or sub-neutralizing antibodies to viral surface proteins that were generated during a previous infection can promote the subsequent entry of viruses into the cell and therefore intensify the inflammatory process during a secondary infection with any antigenic-related virus.6,8
ADE was first described in 1964 by Hawkes, who demonstrated increased infectivity of various arboviruses such as Japanese encephalitis virus, West Nile virus, Murray Valley encephalitis virus, and Murray Valley virus and Getah virus under in vitro conditions. 6 Prior to that, there were also previous reports positing pre-existing non-neutralizing antibodies as responsible for increased infection with various human and animal viruses, including dengue virus (DENV), Zika virus (ZIKV), Ebola virus, human immunodeficiency virus (HIV), Aleutian mink disease parvovirus, Coxsackie B virus, equine infectious anemia virus, feline infectious peritonitis virus, simian hemorrhagic fever virus, caprine arthritis virus, respiratory syndrome virus, and reproductive disease and African swine fever virus. To date, ADE has also been demonstrated with models using monoclonal antibodies and in vitro models of polyclonal sera using cells expressing the Fc receptor, including K562 and U937 cell lines, as well as primary human monocytes, macrophages, and dendritic cells. 44
Molecular mechanism of ADE
In order to clearly understand ADE, it has been broadly categorized into two different mechanisms; when the specific antibody enhances viral entry into host monocytes/macrophages and granulocytes or when it promotes viral infection in cells through interaction with FcR and/or complement receptor. Although these mechanisms are not mutually exclusive, their classification was proposed in order to understand the biological process involved at the molecular level.8,45
ADE via enhanced infection
As mentioned earlier, FcRs are mostly expressed by immune cells and are receptors directed to Fc portion of an antibody. In ADE, via enhanced infection, non-neutralizing or sub-neutralizing antibodies bind to the viral surface and traffic virions directly to macrophages, this complex is internalized by Fc-receptor-bearing cells, including monocytes/macrophages and dendritic cells and subsequently leads to the phosphorylation of Syk and PI3K that triggers signaling for FcγR-mediated phagocytosis. Alternatively, activating FcγR can concentrate immune complexes on the surface of the cell. The virion can then bind to its receptor to enter the cell via receptor-mediated endocytosis. These processes culminate in an increased virus load and disease severity (Figure 2(b)).8,44,46
It is also worth mentioning that this mechanism can be abrogated in the absence of the Fc receptor. The activation of Fc receptors triggers signaling molecules that also induce IFN-stimulated gene (ISG) expression, independent of type-I IFN. Because ISGs have powerful antiviral effects, viruses must develop tools to suppress these antiviral responses in target cells for ADE to occur. For example, in DENV infection, the ADE phenomenon requires the binding of DENV to the leukocyte immunoglobulin receptor B1 (LILRB1). As a result, LILRB1 signaling can inhibit the pathway that induces ISG expression.47,48
ADE via enhanced immune activation
The second, recently described and less studied mechanism, through which ADE can occur, is well represented by pathogens that cause respiratory infections. In these conditions, Fc-mediated antibody effector functions are capable of enhancing respiratory disease by initiating a strong immune cascade that results in severe lung pathology (Figure 2(c)). 45
This mechanism can also be induced when virus–antibody C1q complexes promote fusion between the viral capsule and the cell membrane by deposition of C1q and its receptor. This complex binds to the C1q receptor in cells and initiates the intracellular signaling pathway. The classical complement pathway is then initiated, leading to the activation of C3, whose fragment can be covalently linked to the bound antibodies or the surface of the virus particles then favors the binding of the virus and its receptor, as well as the subsequent endocytosis.10,43
Interestingly, another mechanism for the ADE phenomenon that has been rather described in the multisystemic inflammatory syndrome in children is that mediated by mast cells; these cells are capable of degranulating both IgE and IgG antibodies bound to Fc receptors. 49
In this sense, a model of multisystemic inflammatory syndrome in children has been proposed in babies with maternally transferred antibodies against SARS-CoV-2 in which the activation and degranulation of mast cells with SARS-CoV-2 antibodies bound to the Fc receptor lead to an increase in histamine levels. In this model, the binding of the SARS-CoV-2 nucleocapsid to the PTGS2 promoter results in prostaglandin E2 (PGE2) which may be driving overactive mast cells as an alternative mechanism that drives increased histamine levels in older children and adults. 49
The best known so far (but also misunderstood) ADE phenomenon: ADE in DENV infection
The DENV is a mosquito-borne virus of the Flaviviridae family (with four serotypes identified DENV1-4) capable of causing classic dengue (DF), dengue hemorrhagic fever (DHF), and dengue shock syndrome (DSS) showing tropism for monocytes, macrophages, and dendritic cells.42,48,50
There exists no cross-antibody protection for the four serotypes, which means the antibodies induced by each serotype cannot work on others. In the case of a secondary infection, if infected by the virus of same serotype, the antibodies produced in previous infections are capable of effectively neutralizing the virus. On the contrary, these antibodies will not only neutralize viruses, but may also even facilitate viral entry through Fc portions of antibodies and will increase viral load in vivo. 8
According to this hypothesis of ADE, the antibodies produced in a DENV infection can recognize and bind to a different serotype of DENV than that of the primary infection but are not able to neutralize it. Instead, these antibodies facilitate the entry of non-neutralized virus–antibody complexes (immune complexes), primarily through FcγR into phagocytic mononuclear cells (MPCs). 48
The DENV represents the best documented example of clinical ADE via enhanced infection. After ligation of FcR, DENV activates IL-10 production at an early phase of infection. The suppressor activity of IL-10 during ADE infection induces Th2 bias and inhibits the JAK-STAT signaling pathway through the suppressor cytokine signaling system (SOCS). ADE also results in a higher rate of virus internalization by increasing the number of fusions per cell.44,45,51
Since many antibodies to different dengue serotypes are cross-reactive, secondary infections with heterologous strains can lead to increased viral replication and more severe disease. Typically, both DHF and DSS occur in this setting, presenting more severe forms of symptoms, such as thrombocytopenia, fever, and hemorrhagic manifestations. It has also been shown that the presence of these cross-reactivated non-neutralizing antibodies can predispose to more severe disease and even the development of DHF and DSS.45,52
SARS CoV-2 and ADE, what is known and what remains to be known?
Despite all reports generated in recent months in response to the pandemic, there is still no detailed information regarding the mechanism of the ADE that occurs in SARS-CoV-2 infection. One of the best accepted hypotheses so far is that in the SARS-CoV-2 infection, pre-existing CoV-specific antibodies are capable of promoting viral entry into FcR-expressing cells. ADE is mediated by the binding of FcRs, mainly CD32 expressed in different immune cells, including monocytes, macrophages, and B cells. The infection of CD32+ cells is a key step in the development of the COVID-19 and its progression from mild to severe form.53,54
A potential hypothesis states that circulating non-neutralizing antibodies, instead of helping to eliminate circulating SARS-CoV-2, can then bind to viral particles and thus contribute to the worsening of COVID-19 by promoting its Fc-mediated internalization by pulmonary epithelial cells and infiltrating monocytes, as it has been observed in previously mentioned diseases such as SARS-CoV-1. 55
One particularity about this mechanism is that ADE of SARS-CoV does not use endosomal/lysosomal pathway as used by ACE2 during normal virus transport into the cell, but instead it has been described as a possible mechanism for viral entry where non-neutralizing antibodies recognizing the RBD of the S-protein of the coronavirus bind to the Fc receptor and allow virus entry. The non-neutralizing antibodies–Fc receptor complex mimics the cell surface virus receptor and favors virus entry pathways into IgG Fc receptor-expressing cells.6,52
This phenomenon could also explain the observed impairment of immune regulation such as apoptosis of immune cells leading to the development of T-cell lymphopenia, an inflammatory cascade, as well as a storm of cytokines.8,54
An important difference between the ADE phenomenon previously described for DENV and SARS-CoV is that there is no evidence that ADE facilitates the spread of SARS-CoV in infected hosts. Therefore, ADE in this disease would be best described as “ADE of viral entry” which does not necessarily result in a productive viral infection, meaning that ADE of viral entry in vitro does not predict ADE of infection and ADE of disease. 56
Antibodies are capable of promoting virus attachment and entry into the immune cell, where they start to replicate without production of viable virions. This pseudo infection may be due to the inability of macrophages to express the serine proteases necessary for virion activation. For their part, immune complexes (virus–antibody) can promote an infectious process after being internalized through the FcRs. Furthermore, pulmonary epithelial cells have been reported to express high levels of FcγRIIa. The virus introduced into the endosome through this pathway will likely involve TLR3, TLR7, and TLR8 capable of recognizing RNA. SARS-CoV infection by ADE in macrophages leads to elevated production of TNF and IL-6. It was also observed in a murine SARS-CoV model that ADE is associated with a decrease in the levels of the anti-inflammatory cytokines IL-10 and TGFβ and increased levels of the pro-inflammatory chemokines CCL2 and CCL3.7,53,54
ADE in the case of SARS-CoV-2 can occur due to the priming caused by other CoVs, leading to development of non-neutralizing or poorly neutralizing antibodies. It is known that antibodies to the S-proteins of SARS-CoV and SARS-CoV-2—and, to a much lesser extent, MERS-CoV—can cross-react, and both high-potency neutralizing antibodies that also mediate antibody-dependent cytotoxicity and antibody-dependent cellular phagocytosis, as well as non-neutralizing antibodies, can be elicited against conserved S-epitopes. Despite the above, the limited spread of SARS-CoV and MERS-CoV means that it is not feasible that antibodies with cross-reactivity due to another coronavirus infection are the responsible element for the development of ADE, but rather those that were generated during a first infection or after passive immunization.8,57
The ADE hypothesis is further supported by the results of a study on viral kinetics and antibody responses in patients with COVID-19 where it was found that stronger antibody response was associated with delayed viral clearance and increased disease severity. Patients with an elevated IgG response showed only 9% of virus shedding on day 7 after IgG developed. In the case of weak IgG patients, 57% shed the virus. Furthermore, an association was found between a more severe disease phenotype and earlier IgG response, concurrently with IgM and higher IgG antibody titers. 58
The hypotheses regarding ADE are however conflictive and somehow even contradictory. As stated by Jaume et al., it was observed in an in vitro analysis that ADE infection promoted viral gene transcription and the production of viral gene protein synthesis and intermediate species, which can be then recognized by immune sensors and potentiate an immune response. Therefore, proposing a possible participation of immune-mediated enhanced disease during SARS pathogenesis suggests very little clinical significance for this mechanism. In this same study, it was observed in a different cell line, (Raji cells, derived from a Burkitt’s lymphoma patient) that ADE infected cells did not support replication of SARS-CoV-1, ultimately ending in an abortive viral cycle without the detectable release of progeny virus. 59
In addition to the above, recent reports indicate that the percentage of patients with COVID-19 that develop cross-reactive antibodies is significant. In a study by Shrock et al., a serological profile of patients with and without previous COVID-19 infection was performed. In this study, it was found that the studied patients presented cross-reactive antibody titers, and it is suggested that this may have various effects on the disease, from a less severe prognosis when they were able to neutralize the virus to a serious infection when ADE is developed. 60
Another important aspect that needs to be studied further is the relationship between ADE-epitopes. This was previously reported for DENV and ZIKV.61,62 In the case of SARS-CoV-2, this association was reported for the first time in the article by Zhou et al., where monoclonal cells were isolated from memory B cells, later a group of non-overlapping receptor-binding domain was identified. (RBD) epitopes that were directly associated with ADE and favored the entry of the virus into Raji cells via an Fcg receptor-dependent mechanism. 56 This is of utmost importance especially when considering the design of vaccines, which, as mentioned later, must be capable of triggering a strong neutralizing response, which is why the epitopes to which they will be directed must be carefully selected.
Finally, it is also important to take into account that detailed research is lacking to elucidate the possible mechanism of ADE in SARS-CoV-2 infection, mainly due to the fact that the studies carried out at present have been carried out in viral infections (such as DENV) with differences in their pathological mechanisms as well as in animal models (such as the feline infectious peritonitis virus [FIPV]) where mechanisms of pathogenesis in the human host differ among viruses, therefore difficult to translate the mechanisms of infection. 57
ADE as a possible threat to vaccine efficacy
All vaccines have the objective of generating a response from the host against an antigen that is not capable of causing a disease but of provoking a response against that antigen that will be effective in subsequent encounters with it. As we have been discussing, the mechanism of ADE makes vaccine development particularly difficult due to similarity to a natural infection. Vaccines against one specific serotype produce cross-reactive non-neutralizing antibodies against other serotypes, predisposing the enhanced illness in secondary heterotypic infection. 52
The immune mechanisms of this phenomenon involve from ADE of infection to the formation of immune complexes by antibodies, although accompanied by various coordinated cellular responses, such as Th2 T-cell skewing. 63 Another important point to consider is that not only sub-neutralizing or non-neutralizing antibodies are associated with the development of ADE; according to the study by Liu et al., 55 anti-spike IgG (S-IgG), in productively infected lungs, causes severe ALI by skewing inflammation-resolving response.
To avoid the development of ADE, the strategy used in the development of current vaccines was to target the immunodominant epitope, in this case, that corresponds to the S-protein. The S1 subunit presents two highly immunogenic domains, the N-terminal domain (NTD) and the RBD, which are the major targets of polyclonal and monoclonal neutralizing antibodies.64,65 Because the S-proteins of SARS-CoV-2 are accessible and play an essential role in the entry of the virus into the host cell, and therefore the mechanism of infection, they are considered to be prime antibody targets. 66
Understanding the structure of SARS-CoV-2 epitopes, particularly within S, provides essential information for the development of vaccines that favors the production of neutralizing antibodies rather than antibodies that could exacerbate the severity of ADE infection. 60 In general, RNA viruses are known to be highly susceptible to random mutations due to the lack of exonuclease proofreading activity of virus-encoded RNA-dependent RNA polymerases (RdRp) 67 with some exceptions such as Nidovirales order (to which the Coronavirus genus belongs). In SARS-CoV, an exonuclease activity with proofreading function has been described for the nsp14 (ExoN), and a homologue nsp14 protein is found in the SARS-CoV-2 as well. 68
The high error rate and subsequent rapid evolution of virus populations, which could lead to the accumulation of amino acid mutations, could affect the virus’ transmissibility, its cellular tropism, and even its pathogenicity. 69
Although several vaccines have gained (emergence) regulatory approval and are being distributed worldwide, we cannot ignore the possibility that the evolution of the virus, based on natural selection, can directly affect the S-protein to which these vaccines are directed, and therefore the newly mutated virus can escape antibody-mediated protection induced by previous infection or vaccination. 70
Amino acid sequences of SARS-CoV-2 are available from NCBI GenBank and by the Global Initiative on Sharing All Influenza Data (GISAID). The first complete genome sequence of SARS-CoV-2 was released on NCBI GenBank (NC 045512.2). 67 According to these reported sequences, the linear genome of the SARS-CoV-2 virus is 29,903 bases long and houses 25 genes. 71 To date, 4150 mutations have been identified in the S-gene of SARS-CoV-2 isolated from humans, resulting in 1246 changes in amino acids, including 187 RBD substitutions compared to the reference genome. 72
The main variants identified that seem to have high relevance in the immunogenicity of the virus are D614G, N501Y, and E484K mutations of the RBD.73–76
The D614G mutation in protein S represents a change from nucleotide A to G at position 23,403 in the first Wuhan reference strain. The D614G change is commonly detected along with three other mutations: a C to T change in the UTR 5 ‘ (position 241 relative to the Wuhan reference sequence), a silent mutation from C to T at position 3037, and a C-to-T mutation at position 14,408 that results in an amino acid change in the RdRp P323L. This, comprised the four aforementioned mutations, represented the dominant global form as of May (78% of a total of 12,194 sequences). 73 The D614G mutation has been reported as capable of improving the replication capacity of SARS-CoV-2 in the upper respiratory tract through increased virion infectivity, this was demonstrated in the human lung cell line Calu-3 and the primary tissues of the human upper respiratory tract. 73
It was also observed that patients infected with the G614 variant of the virus developed higher levels of viral RNA in nasopharyngeal smears than those with the D614 virus but did not develop a more severe disease. This suggests that despite affecting the replication capacity of the virus, this mutation did not influence the severity of the infection. 77
The N501Y variant was identified in the UK as VUI-2020/01 or lineage B.1.1.7. This lineage is composed of 14 defining mutations in protein S. This variant has a mutation in the RBD of the peak protein at position 501, where the amino acid asparagine (N) has been replaced by tyrosine (Y). The N501Y mutation is one of the six key contact residues within the RBD. 78
This change in different fundamental residues in the binding site could affect the fusion of the host cells–virus and, therefore, the infectivity of the virus. 79 As of December 28, 2020, this variant accounted for approximately 28% of cases of SARS-CoV-2 infection in England. 74
The E484K mutation in the S-protein of the virus has been identified in the South African (B.1.351) and Brazilian (B.1.1.28) variants and has been reported to be an escape mutation from the immune response. 80
This variant consists of a change in codon 484 in that of the RBD where a negatively charged amino acid (E, glutamic acid) is substituted with a positively charged amino acid (K, lysine). 81
Due to the location of this mutation, like the other variants, it has been directly associated with changes in the mechanism of infection of the virus and even on the efficacy of the immune response of the organism or that induced by a vaccine to the virus. 80 Studies have also shown that the presence of this variant directly affects the average binding of convalescent sera (>10 times) reducing the neutralization activity of some individuals. 75
Recently, the BNT162b2 nucleoside modified RNA vaccine encoding the full-length SARS-CoV-2 protein (S) was reported to be effective in inducing neutralizing geometric mean titers of antibodies against SARS-CoV-2 virus constructs containing key peak mutations of the newly emerging UK (UK) and South African (SA) variants: N501Y from the UK and South Africa; Deletion 69/70 + N501Y + D614G from the UK; and E484K + N501Y + D614G de SA, thus suggesting that the efficacy of this vaccine is not significantly affected by these variants. 82
Recently, the delta variant (B.1.617.2) was described, which is characterized by mutations in the peak protein P681R, T19R, D614G, L452R, T478K, Δ157-158, and D950N, first detected in India in December 2020. 83 According to what is believed, these mutations directly affect key antigenic regions of RBD. This variant also appears to cause mutations at sites that trigger an increase in viral replication and therefore an increase in viral load. 84 This variant and its rapid transmission capacity represent an imminent threat to the population and a concern about the effectiveness of vaccines. In this sense, in the study by Lopez-Bernal et al., it was reported that the effectiveness after a dose of vaccine (BNT162b2 or ChAdOx1 nCoV-19) was lower among people with the delta variant (30.7%) than among those with the alpha variant (48.7%). With the BNT162b2 vaccine, the effectiveness of two doses was 93.7% among people with the alpha variant and 88.0% among people with the delta variant. With the ChAdOx1 nCoV-19 vaccine, the two-dose efficacy was 74.5% among people with the alpha variant and 67.0% among people with the delta variant. 84
In addition to this, there are reports regarding the kinetics of natural immunity in patients who had COVID-19. In a study, 85 the humoral response was evaluated in a total of 76 patients (IgM and IgG antibodies that recognized the nucleocapsid protein or the RBD of the S-protein). In these patients 1 year after infection, approximately 90% of recovered patients still had detectable SARS-CoV-2-specific IgG antibodies recognizing N and RBD-S. However, when evaluating the neutralizing capacity, it was only detected in ∼43% of patients. 85
In addition to concerns regarding natural immunity, there are also reports about the duration of the humoral immune response in response to a vaccine. In a study in health personnel vaccinated with BNT162b2, it was observed that the antibody response was greater in seropositive participants compared to seronegative participants. In both seropositive and seronegative subjects, a significant decrease in antibodies was observed at 3 months compared to maximum response. 86 Similar results were found by our work group in the study by Morales-Nuñez et al., where it was observed that after the second dose with this same vaccine, individuals developed antibodies with high neutralizing capacity. 87 In a study by Pegu et al., 2021, the efficacy of the immune response generated by the mRNA-1273 vaccine was evaluated, in this work the impact of the variants B.1.1.7 (Alpha), B.1.351 (Beta), P.1 was also evaluated (Gamma), B.1.429 (Epsilon), B.1.526 (Iota) for SARS-CoV-2, and B.1.617.2 (Delta) on binding, neutralization, and ACE2-competing antibodies elicited by this vaccine for 7 months. The results of this study turned out to be interesting because all included individuals responded to all variants. Binding and functional antibodies against variants persisted in most subjects, albeit at low levels, for 6 months after the primary series of mRNA-1273 vaccine. 88
The imminent risk that may be triggered by a vaccine-mediated antibody response is that the mechanism of ADE occurs and places vaccinated individuals at greater risk of a more severe disease phenotype compared to unvaccinated individuals. Closely monitoring of these mutations is essential for the scientists in charge of the design and development of vaccines to make the necessary modifications that go hand in hand with the high mutation rate of SARS-CoV-2. 63
The light at the end of the tunnel; an inhibitor as a possible therapeutic alternative
As described above in the presence of cross-reactive antibodies (responsible for the ADE phenomenon), the entry of the virus is promoted in monocytes/macrophages through the FcR. Once inside the cell, the viruses are replicated and released in large quantities after escaping the immune response. The exacerbated activation of macrophages and mass liberation of cytokines support a hypothesis that states that the so-called cytokine storm is the secondary event of the activation of macrophages, mainly mediated by the ADE phenomenon, reason why its specific blockade will provide therapeutic potentials for patients suffering from severe COVID-19. 89
In this context, it has been stated that the mammalian Target of Rapamycin (mTOR) is one of the main signaling pathways involved in the exacerbated immune response triggered by SARS-CoV2. 90 mTOR is a serine-threonine kinase family protein, a key regulator in protein synthesis, and cellular metabolism that forms two major complexes, mTORC1 with Raptor and mTORC2 with Rictor and plays a pivotal role in cell proliferation and cellular metabolism; therefore, inhibition of mTOR has shown to suppress virus growth and replication. 91
In this regard, in a recent study, a specific set of biological pathways was described in the primary human pulmonary epithelium of SARS-COV-2 infection, among them the mTOR signaling pathway was identified. 92 It has also been stated that the mTOR pathway plays an important role in B‐cell development; mTORC1 controls BCL6 expression and controlling the fate of B cells in the germinal center reaction, therefore contributing in an essential way to the development of ADE by favoring the production of cross-reactive or sub-neutralizing antibodies. 89
These findings propose that selective inhibition of mTOR by an inhibitory agent, such as rapamycin, could have detrimental effects over memory B cell activation and therefore beneficial effects over the characteristic immune response of COVID-19 92
The mechanism of action of rapamycin consists of its ability to bind to the FK506 Immunophilin-binding protein (FKBP12A) and to inhibit the activity of mTORC1 as well as to interrupt the interaction between Raptor and mTOR. The inhibition of mTORC1 by rapamycin then leads to autophagy of infected cells and inhibition of translation of SARS‐CoV‐2 viral polymerase and structural proteins. 90
Overall, it is suggested that the antiviral action of rapamycin, together with its immunomodulatory potential that reduces the excessive production of pro-inflammatory cytokines, would justify clinical studies in patients with COVID-19.90,91
Conclusions
The outbreak and rapid spread of SARS-CoV-2 are a health threat with unprecedented consequences throughout the world. Considering the great economic and health burden of the COVID-19 pandemic, any means to improve the condition of patients, accelerate their recovery, and reduce the risk of deterioration and death would be considered of significant clinical and economic importance. With respect to the immune response generated by the host, the specific neutralizing antibodies generated against the virus are considered essential in the control of virus infections in various ways. However, in some cases, the presence of specific antibodies can be beneficial for the virus. This activity known as antibody-dependent enhancement (ADE) of virus infection enhances virus entry and in some cases virus replication into host cells through interaction with Fc and/or complement receptors. It has been also reported in data from previous CoV research studies that ADE may play a role in the virus’s pathology.
Even though several vaccines have been approved from regulatory bodies under emergency conditions and are distributed worldwide, we cannot rule out the possibility that the evolution of the virus can directly affect its targets, and therefore, the newly mutated virus can escape antibody-mediated protection induced by previous infection or vaccination.
If the vaccines are not capable of generating neutralizing antibodies against the possible mutagenic variants to mount a response, the result may lead to the generation of sub-neutralizing antibodies that will even be capable of facilitating uptake by macrophages that express FcR, with the subsequent stimulation of macrophages and production of pro-inflammatory cytokines.
One advantage of the current pandemics is the unprecedented availability of scientific and technological means to face COVID-19, on these bases, careful design and testing of vaccines will be necessary to evaluate which viral mutations can escape from antibodies-mediated neutralization as well as which one significantly affects the efficacy of the currently approved vaccines.Go to:
Acknowledgments
The figures were created with BioRender.comGo to:
Appendix
Abbreviations
ACE2Angiotensin-converting enzyme 2ADEAntibody-dependent enhancementCoVsCoronavirusesCOVID-19Coronavirus-19DENVDengue virusDFDengue feverDHFDengue hemorrhagic feverDSSDengue shock syndromeEREndoplasmic reticulumFcRsFc receptorsGISAIDGlobal Initiative on Sharing All Influenza DataHIVHuman immunodeficiency virusIDIntradermalIFNInterferonILInterleukinIMIntramusculariNOSInducible nitric oxide synthaseIRFInterferon regulatory factorISGIFN stimulated geneLILRB1Leukocyte immunoglobulin-like receptor B1MERSMiddle East respiratory syndromeMHCMajor histocompatibility complexMPCsMononuclear phagocytic cellsmTORMammalian Target of RapamycinNF-KBNuclear factor kBNKsNatural killer cellsNTDN-terminal domainORFOpen reading framesPGE2Prostaglandin E2PRRsPattern recognition receptorsRBDReceptor-binding domainRdRpRNA-dependent RNA polymeraseRLRRIG-I-like receptorsRNARibonucleic acidROSReactive oxygen speciesRSVRespiratory Syncytial VirusSOCSSuppressor of cytokine signallingThT helper cellTLRToll-like receptorTMPRRS2Serine protease transmembrane type 2TNF-αTumor necrosis factorWHOWorld Health OrganizationZIKVZika virusGo to:
Footnotes
Author contributions: Gabriela Athziri Sánchez-Zuno, Mónica Guadalupe Matuz-Flores, and José Francisco Muñoz-Valle conceived, drafted, and finalized the manuscript.José Francisco Muñoz-Valle, Francisco Javier Turrubiates-Hernández, and Guillermo González-Estevez critically reviewed the draft of the manuscript and approved the final version.All the authors contributed significantly and agreed to the published version of the manuscript.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Council of Science and Technology (CONACYT Ciencia Básica grant number A1-S-8774) and the Universidad de Guadalajara through Fortalecimiento de la Investigación y el Posgrado 2020.Go to:
ORCID iDs
Francisco Javier Turrubiates-Hernández https://orcid.org/0000-0001-9637-168X
José Francisco Muñoz-Valle https://orcid.org/0000-0002-2272-9260Go to:
References
1. National Center for Immunization and Respiratory Diseases (NCIRD), Division of Viral Diseases (2020) Scientific Brief: SARS-CoV-2 Transmission. In CDC COVID-19 Science Briefs Atlanta (GA): Centers for Disease Control and Prevention (US). [Google Scholar]
2. Coronavirus Disease (COVID-19) Situation reports. [online] Available at: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed April 2021).
3. Baloch S, Baloch MA, Zheng T, et al. (2020) The Coronavirus Disease 2019 (COVID-19) pandemic. Tohoku J Exp Med 250: 271–278. doi:10.1620/tjem.250.271. [PubMed] [CrossRef] [Google Scholar]
4. Zhou Y, Chi J, Lv W, et al. (2021) Obesity and diabetes as high‐risk factors for severe coronavirus disease 2019 (Covid‐19). Diabetes Metab Res Rev 37(2): e3377. doi:10.1002/dmrr.3377. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
5. Mehta P, McAuley DF, Brown M, et al. (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395: 1033–1034. doi:10.1016/S0140-6736(20)30628-0. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
6. Karthik K, Senthilkumar TMA, Udhayavel S, et al. (2020) Role of antibody-dependent enhancement (ADE) in the virulence of SARS-CoV-2 and its mitigation strategies for the development of vaccines and immunotherapies to counter COVID-19. Hum Vaccines Immunother 16(12): 3055–3060. doi:10.1080/21645515.2020.1796425. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
7. Peron JPS, Nakaya H. (2020) Susceptibility of the elderly to SARS-CoV-2 infection: ACE-2 overexpression, shedding, and antibody-dependent enhancement (ADE). Clinics 75, e1912. doi:10.6061/clinics/2020/e1912. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
8. Wen J, Cheng Y, Ling R, et al. (2020) Antibody-dependent enhancement of coronavirus. Int J Infect Dis 100: 483–489. doi:10.1016/j.ijid.2020.09.015. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
9. Ujike M, Taguchi F. (2015) Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses 7: 1700–1725. doi:10.3390/v7041700. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
10. Comas-Garcia M. (2019) Packaging of genomic RNA in positive-sense single-stranded RNA viruses: a complex story. Viruses 11: 253. doi:10.3390/v11030253. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
11. Kirchdoerfer RN, Cottrell CA, Wang N, et al. (2016) Pre-fusion structure of a human coronavirus spike protein. Nature 531; 118–121. doi:10.1038/nature17200. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
12. Song W, Gui M, Wang X, et al. (2018) Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLOS Pathog 14: e1007236. doi:10.1371/journal.ppat.1007236. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
13. National Center for Immunization and Respiratory Diseases (NCIRD), Division of Viral Diseases (2020) Science brief: SARS-CoV-2 and surface (Fomite) transmission for indoor community environments. In CDC COVID-19 Science Briefs. Atlanta (GA): Centers for Disease Control and Prevention (US). [Google Scholar]
14. Li W, Moore MJ, Vasilieva N, et al. (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450–454. doi:10.1038/nature02145. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
15. HCA Lung Biological Network. Sungnak W, Huang N, Bécavin C, et al. (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 26: 681–687. doi:10.1038/s41591-020-0868-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
16. Fung TS, Liu DX. (2019) Human coronavirus: host-pathogen interaction. Annu Rev Microbiol 73: 529–557. doi:10.1146/annurev-micro-020518-115759. [PubMed] [CrossRef] [Google Scholar]
17. Hur S. (2019) Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37: 349–375. doi:10.1146/annurev-immunol-042718-041356. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
18. Young RE, Thompson RD, Larbi KY, et al. (2004) Neutrophil Elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. J Immunol 172: 4493–4502. doi:10.4049/jimmunol.172.7.4493. [PubMed] [CrossRef] [Google Scholar]
19. Liu S, Su X, Pan P, et al. (2016) Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep 6: 37252. doi:10.1038/srep37252. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
20. Lokugamage KG, Hage A, de Vries M, et al. (2020) Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. J Virol 94(23): e01410–e01420. [PMC free article] [PubMed] [Google Scholar]
21. Stanifer ML, Kee C, Cortese M, et al. (2020) Critical role of type III interferon in controlling SARS-CoV-2 infection, replication and spread in primary human intestinal epithelial cells. Cell Rep 32(1): 107863. [PMC free article] [PubMed] [Google Scholar]
22. Channappanavar R, Fehr AR, Zheng J, et al. (2019) IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest 129: 3625–3639. doi:10.1172/JCI126363. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
23. Kaneko N, Kuo H-H, Boucau J, et al. (2020) Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell 183: 143–157.e13. doi:10.1016/j.cell.2020.08.025. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
24. Hadjadj J, Yatim N, Barnabei L, et al. (2020) Impaired type I interferon activity and exacerbated inflammatory responses in severe covid-19 patients. Science 369(6504): 718–724. [PMC free article] [PubMed] [Google Scholar]
25. Knoops K, Kikkert M, Van den Worm SHE, et al. (2008) SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol 6: e226. doi:10.1371/journal.pbio.00602
26. [PMC free article] [PubMed] [CrossRef] [Google Scholar]26. Chen Y, Cai H, Pan J, et al. (2009) Functional screen reveals SARS coronavirus nonstructural Protein Nsp14 as a Novel Cap N7 methyltransferase. Proc Natl Acad Sci Unit States Am 106: 3484–3489. doi:10.1073/pnas.0808790106. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
27. Bouvet M, Debarnot C, Imbert I, et al. (2010) Vitro reconstitution of SARS-coronavirus MRNA cap methylation. PLoS Pathog 6: e1000863. doi:10.1371/journal.ppat.1000863. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
28. Jansen JM, Gerlach T, Elbahesh H, et al. (2009) Influenza virus-specific CD4+ and CD8+ T cell-mediated immunity induced by infection and vaccination. J Clin Virol 119: 44–52. doi:10.1016/j.jcv.2019.08.009. [PubMed] [CrossRef] [Google Scholar]
29. Long Q-X, Liu B-Z, Deng H-J, et al. (2020) Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med 26: 845–848. doi:10.1038/s41591-020-0897-1. [PubMed] [CrossRef] [Google Scholar]
30. Wang F, Nie J, Wang H, et al. (2020) Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis 221: 1762–1769. doi:10.1093/infdis/jiaa150. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
31. Fung S-Y, Yuen K-S, Ye Z-W, et al. (2020) A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg Microb Infect 9: 558–570, doi:10.1080/22221751.2020.1736644. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
32. Welsh RM, Bahl K, Marshall HD, et al. (2012) Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog 8: e1002352. doi:10.1371/journal.ppat.1002352. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
33. Sallard E, Lescure F-X, Yazdanpanah Y, et al. (2020) Type 1 interferons as a potential treatment against COVID-19. Antivir Res 178: 104791. doi:10.1016/j.antiviral.2020.104791. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
34. Liu J, Li S, Liu J, et al. (2020) Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 55: 102763. doi:10.1016/j.ebiom.2020.102763. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
35. Qin C, Zhou L, Hu Z, et al. (2020) Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis 71: 762–768. doi:10.1093/cid/ciaa248. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
36. Hu B, Huang S, Yin L. (2021) The cytokine storm and COVID‐19. J Med Virol 93(1): 250–256. doi:10.1002/jmv.26232. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
37. Zhu Z, Chakraborti S, He Y, et al. (2007) Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc Natl Acad Sci Unit States Am 104: 12123–12128. doi:10.1073/pnas.0701000104. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
38. Temperton NJ, Chan PK, Simmons G, et al. (2007) Longitudinally profiling neutralizing antibody response to SARS coronavirus with pseudotypes. Emerg Infect Dis 11: 411–416. doi:10.3201/eid1103.040906. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
39. Yuchun N, Guangwen W, Xuanling S, et al. (2004) Neutralizing antibodies in patients with severe acute respiratory syndrome-associated coronavirus infection. J Infect Dis 190: 1119–1126. doi:10.1086/423286. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
40. Ni L, Ye F, Cheng M-L, et al. (2020) Detection of SARS-CoV-2-specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity 52: 971–977.e3. doi:10.1016/j.immuni.2020.04.023. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
41. Takada A, Kawaoka Y. (2003) Antibody-dependent enhancement of viral infection: molecular mechanisms andin vivo implications. Rev Med Virol 13: 387–398. doi:10.1002/rmv.405. [PubMed] [CrossRef] [Google Scholar]
42. Cloutier M, Nandi M, Ihsan AU, et al. (2020) ADE and hyperinflammation in SARS-CoV2 infection- comparison with dengue hemorrhagic fever and feline infectious peritonitis. Cytokine 136: 155256. doi:10.1016/j.cyto.2020.155256. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
43. Kulkarni R. (2020) Antibody-dependent enhancement of viral infections. In: Bramhachari PV. (ed) Dynamics of Immune Activation in Viral Diseases. Singapore: Springer Singapore. pp. 9–41. ISBN 9789811510441. [Google Scholar]
44. Khandia R, Munjal A, Dhama K, et al. (2018) Modulation of Dengue/Zika virus pathogenicity by antibody-dependent enhancement and strategies to protect against enhancement in zika virus infection. Front Immunol 9: 597. doi:10.3389/fimmu.2018.00597. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
45. Lee WS, Wheatley AK, Kent SJ, et al. (2020) Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol 5: 1185–1191. doi:10.1038/s41564-020-00789-5. [PubMed] [CrossRef] [Google Scholar]
46. Gan ES, Ting DHR, Chan KR. (2017) The mechanistic role of antibodies to dengue virus in protection and disease pathogenesis. Expert Rev Anti Infect Ther 15: 111–119. doi:10.1080/14787210.2017.1254550. [PubMed] [CrossRef] [Google Scholar]
47. Wan Y, Shang J, Sun S, et al. (2020) Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J Virol 94: e02015–e02019. doi:10.1128/JVI.02015-19. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
48. Langerak T, Mumtaz N, Tolk VI, et al. (2019) The possible role of cross-reactive dengue virus antibodies in zika virus pathogenesis. PLOS Pathog 15: e1007640. doi:10.1371/journal.ppat.1007640. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
49. Ricke DO, et al. (2021) Two different antibody-dependent enhancement (ADE) risks for SARS-CoV-2 antibodies. Front Immunol 12: 640093. doi:10.3389/fimmu.2021.640093. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
50. Pang X, Zhang R, Cheng G. (2017) Progress towards understanding the pathogenesis of dengue hemorrhagic fever. Virol Sin 32: 16–22. doi:10.1007/s12250-016-3855-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
51. Halstead SB, Mahalingam S, Marovich MA, et al. (2010) Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis 10: 712–722. doi:10.1016/S1473-3099(10)70166-3. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
52. Ulrich H, Pillat MM, Tárnok A, et al. (2020) Dengue fever, COVID ‐19 (SARS‐CoV ‐2), and antibody‐dependent enhancement (ADE): a perspective. Cytometry 97: 662–667. doi:10.1002/cyto.a.24047. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
53. Iwasaki A, Yang Y. (2020) The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol 20: 339–341. doi:10.1038/s41577-020-0321-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
54. Zaichuk TA, Nechipurenko YD, Adzhubey AA, et al. (2020) The challenges of vaccine development against betacoronaviruses: antibody dependent enhancement and sendai virus as a possible vaccine vector. Mol Biol 54(6): 922–938. doi:10.1134/S0026893320060151. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
55. Liu L, Wei Q, Lin Q, et al. (2019) Anti–spike IgG causes severe acute lung injury by skewing macrophage responses during Acute SARS-CoV infection. JCI Insight 4: e123158. doi:10.1172/jci.insight.123158. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
56. Zhou Y, Liu Z, Li S, et al. (2021) Enhancement versus neutralization by SARS-CoV-2 antibodies from a convalescent donor associates with distinct epitopes on the RBD. Cell Rep 34: 108699. doi:10.1016/j.celrep.2021.108699. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
57. Arvin AM, Fink K, Schmid MA, et al. (2020) A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 584: 353–363. doi:10.1038/s41586-020-2538-8. [PubMed] [CrossRef] [Google Scholar]
58. Fierz W, Walz B. (2020) Antibody dependent enhancement due to original antigenic sin and the development of SARS. Front Immunol 11: 1120. doi:10.3389/fimmu.2020.01120. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
59. Jaume M, Yip MS, Cheung CY, et al. (2011) Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a PH- and cysteine protease-independent Fc R pathway. J Virol 85: 10582–10597. doi:10.1128/JVI.00671-11. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
60. Shrock E, Fujimura E, Kula T, et al. (2020) Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 370(6520): eabd4250. doi:10.1126/science.abd4250. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
61. Shukla R, Ramasamy V, Shanmugam RK, et al. (2020) Antibody-dependent enhancement: a challenge for developing a safe dengue vaccine. Front Cell Infect Microbiol 10: 572681. doi:10.3389/fcimb.2020.572681. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
62. Cui G, Si L, Wang Y, et al. (2021) Antibody‐dependent enhancement (ADE) of dengue virus: identification of the key amino acid that is vital in denv vaccine research. J Gene Med 23(2): e3297. doi:10.1002/jgm.3297. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
63. Cardozo T, Veazey R. (2021) Informed consent disclosure to vaccine trial subjects of risk of COVID‐19 vaccines worsening clinical disease. Int J Clin Pract 75(3): e13795. doi:10.1111/ijcp.13795. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
64. Andreano E, Piccini G, Licastro D, et al. (2020) SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv, in press. [PMC free article] [PubMed] [Google Scholar]
65. Letko M, Marzi A, Munster V. (2020) Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol 5: 562–569. doi:10.1038/s41564-020-0688-y. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
66. Weisblum Y, Schmidt F, Zhang F, et al. (2020) Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 9: e61312. doi:10.7554/eLife.61312. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
67. Chen J, Wang R, Wang M, et al. (2020) Mutations strengthened SARS-CoV-2 infectivity. J Mol Biol 432: 5212–5226. doi:10.1016/j.jmb.2020.07.009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
68. Pachetti M, Marini B, Benedetti F, et al. (2020) Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med 18: 179. doi:10.1186/s12967-020-02344-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
69. Giovanetti M, Benedetti F, Campisi G, et al. (2021) Evolution patterns of SARS-CoV-2: snapshot on its genome variants. Biochem Biophys Res Commun 538: 88–91. doi:10.1016/j.bbrc.2020.10.102. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
70. Jangra S, Ye C, Rathnasinghe R, et al. (2021) The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. medRxiv, in press. [Google Scholar]
71. Nagy Á, Pongor S, Győrffy B. (2021) Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int J Antimicrob Agents 57: 106272. doi:10.1016/j.ijantimicag.2020.106272. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
72. Liu Z, VanBlargan LA, Bloyet L-M, et al. (2021) Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 29: 477–488.e4. doi:10.1016/j.chom.2021.01.014. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
73. Korber B, Fischer WM, Gnanakaran S, et al. (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182: 812–827.e19. doi:10.1016/j.cell.2020.06.043. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
74. Davies NG, Abbott S, Barnard RC, et al. (2020) Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372(6538): eabg3055. [PMC free article] [PubMed] [Google Scholar]
75. Greaney AJ, Loes AN, Crawford KHD, et al. (2021) Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. Cell Host Microbe 29(3): 463–476.e6. [PMC free article] [PubMed] [Google Scholar]
76. National Institute of Infectious Diseases , JAPAN (2021) Brief report: new variant strain of SARS-CoV-2 identified in travelers from Brazil. January 12, 2021. [Online] Available at: https://www.niid.go.jp/niid/en/2019-ncov-e/10108-covid19-33-en.html (accessed September 2021).
77. Plante JA, Liu Y, Liu J, et al. (2021) Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592: 116–121. doi:10.1038/s41586-020-2895-3. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
78. Erol A. (2021) Are the emerging SARS-COV-2 mutations friend or foe? Immunol Lett 230: 63–64. doi:10.1016/j.imlet.2020.12.014. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
79. Ali F, Kasry A, Amin M. (2021) The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov 10: 100086. doi:10.1016/j.medidd.2021.100086. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
80. Wise J. (2021) Covid-19: the E484K mutation and the risks it poses. BMJ 372: n359. doi:10.1136/bmj.n359. [PubMed] [CrossRef] [Google Scholar]
81. da Silva Francisco R, Jr, Benites LF, Lamarca AP, et al. (2021) Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 Co-infection events by two different lineages in Rio Grande Do Sul, Brazil. Virus Res 296: 198345. doi:10.1016/j.virusres.2021.198345. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
82. Xie X, Liu Y, Liu J, et al. (2021) Neutralization of SARS-CoV-2 Spike 69/70 Deletion, E484K, and N501Y Variants by BNT162b2 Vaccine-Elicited Sera. Nat Med 27(4): 620–621. [PubMed] [Google Scholar]
83. European Centre for Disease Prevention and Control (2021) Emergence of SARS-CoV-2 B.1.617 variants in India and situation in the EU/EEA– 11 May 2021. Stockholm: ECDC [Google Scholar]
84. Lopez Bernal J, Andrews N, Gower C, et al. (2021) Effectiveness of Covid-19 vaccines against the B.1.617.2 (Delta) variant. N Engl J Med 385: 585–594. doi:10.1056/NEJMoa2108891. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
85. Xiang T, Liang B, Fang Y, et al. (2021) Declining levels of neutralizing antibodies against SARS-CoV-2 in convalescent COVID-19 patients one year post symptom onset. Front Immunol 12: 708523. doi:10.3389/fimmu.2021.708523. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
86. Favresse J, Bayart J-L, Mullier F, et al. (2021) Antibody titres decline 3-month post-vaccination with BNT162b2. Emerg Microb Infect 10: 1495–1498. doi:10.1080/22221751.2021.1953403. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
87. Morales-Núñez JJ, Muñoz-Valle JF, Meza-López C, et al. (2021) Neutralizing antibodies titers and side effects in response to BNT162b2 vaccine in healthcare workers with and without prior SARS-CoV-2 infection. Vaccines 9: 742. doi:10.3390/vaccines9070742. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
88. Pegu A, O’Connell S, Schmidt SD, et al. (2021) Durability of MRNA-1273 vaccine–induced antibodies against SARS-CoV-2 variants. Science 2021: eabj4176. doi:10.1126/science.abj4176. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
89. Zheng Y, Li R, Liu S. (2020) Immunoregulation with MTOR inhibitors to prevent COVID‐19 severity: a novel intervention strategy beyond vaccines and specific antiviral medicines. J Med Virol 92: 1495–1500. doi:10.1002/jmv.26009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
90. Karam BS, Morris RS, Bramante CT, et al. (2021) MTOR inhibition in COVID‐19: a commentary and review of efficacy in RNA viruses. J Med Virol 93: 1843–1846. doi:10.1002/jmv.26728. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
91. Ramaiah MJ. (2020) MTOR inhibition and P53 activation, MicroRNAs: the possible therapy against pandemic COVID-19. Gene Rep 20: 100765. doi:10.1016/j.genrep.2020.100765. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
92. Fagone P, Ciurleo R, Lombardo SD, et al. (2020) Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun Rev 19: 102571. doi:10.1016/j.autrev. 2020. 102571. [PMC free article] [PubMed] [CrossRef] [Google Scholar]