
Sergey Knyazev, Karishma Chhugani, Varuni Sarwal, Ram Ayyala, Harman Singh, Smruthi Karel O. Wertheim, Braden T. Tierney, Charles Y. Chiu, Ren Sun, Aiping Wu, Malak S. Abedalthagafi, Victoria M. Pak, Shivashankar H. Nagaraj, Adam L. Smith, Pavel Skums, Bogdan Pasaniuc, Andrey Komissarov, Serghei Mangul
Nature Methods volume 19, pages374–380 (2022) Cite this article 9048 Accesses,17 Citations, 41 Altmetric, Metrics details
During the COVID-19 pandemic, genomics and bioinformatics have emerged as essential public health tools. The genomic data acquired using these methods have supported the global health response, facilitated the development of testing methods and allowed the timely tracking of novel SARS-CoV-2 variants. Yet the virtually unlimited potential for rapid generation and analysis of genomic data is also coupled with unique technical, scientific and organizational challenges. Here, we discuss the application of genomic and computational methods for efficient data-driven COVID-19 response, the advantages of the democratization of viral sequencing around the world and the challenges associated with viral genome data collection and processing.
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly contagious pathogen that caused the COVID-19 pandemic, which reached an unprecedented scale of infection not seen since the influenza pandemic of 1918–1919. Within a month of its first reported case in Wuhan, China, in December 2019, the virus had spread to many regions within China as well as in several neighboring countries, including Thailand, Korea and Japan. As international flights continued to operate, SARS-CoV-2 rapidly spread to Europe and North America1.
During this time, it became clear that the genomic toolkits are essential for public health decision-making, including testing for COVID-19, monitoring for emergence of new virus variants with altered biological or immunological properties, identification of at-risk individuals and informing of epidemiological models that describe outbreaks in communities2. This has allowed the observation of SARS-CoV-2 genome evolution in almost real time and the rapid tracking of SARS-CoV-2 genetic lineages and variants of interest and concern (VOIs, VOCs), which in turn have facilitated the development of clinical tests for SARS-CoV-2 and the prediction of vaccine efficacy against viral variants3,4. However, to reach the full potential of genomic data for future public health surveillance and outbreak response, we believe it is necessary to expand and coordinate best practices in genomics and bioinformatics that have now been field tested during the COVID-19 response5. Herein, we discuss the genomic techniques and corresponding bioinformatics algorithms that are addressing many of the pressing public health issues associated with COVID-19.
Genomics-based methods enabled early warnings of COVID-19 pandemic
As a local team of health professionals was investigating a small local outbreak of pneumonia consisting of the first 59 suspected cases from Wuhan in December 2019, they quickly discovered that they were dealing with a novel virus of unknown origin6. This rapid discovery was made possible by modern robust and accurate genomic and bioinformatic tools that, although now used routinely, did not exist a couple of decades ago. By 30 January 2020, when the World Health Organization (WHO) declared a Public Health Emergency of International Concern (PHEIC), 339 SARS-CoV-2 genomes had already been sequenced and characterized1.
To investigate the newly emerging outbreak, scientists in China performed whole-genome sequencing of specimens, followed by de novo assembly and end-mapping to annotate the complete 29,903-nucleotide-long SARS-CoV-2 genome. Bioinformatics analysis revealed that the genome organization of SARS-CoV-2 was consistent with a single-stranded, positive-sense RNA virus from the genus Betacoronavirus7. Additionally, sequence alignment tools including BLAST8 were used to search for related species of the newly discovered virus in the NCBI GenBank database, revealing alarming similarities to SARS-CoV (SARS-CoV-1), as well as a much higher similarity with Betacoronavirus from bats, suggestive of a zoonotic origin for the virus. Some SARS-CoV-2 genome fragments, in addition, have highest similarity to the corresponding fragments from pangolins, which suggests that recombination events between strains may have occurred during the virus’ evolution. Subsequent analyses that included additional sarbecovirus genomes from bats and pangolins further scrutinized the evolution and recombination history of these viruses, finding that the lineage that gave rise to SARS-CoV-2 had been probably circulating unnoticed in bats for decades9,10.
Genomics-based methods shaped the effective COVID-19 response
Once the SARS-CoV-2 genome was sequenced, the authors immediately publicly deposited the genome in GenBank7,11. This timely open-access release of the virus genome sequence was a laudable decision that allowed informed scientific analyses and pandemic preparation to begin immediately.
As the pandemic progressed, the increased availability of modern sequencing technologies prompted the collection of SARS-CoV-2 viral genomic data on an unprecedented scale. Within a month, on average about 1,300 genomes were being submitted per day. Within six months of start of the pandemic (by May 2020), GISAID had 110,000 full-length SARS-CoV-2 genome sequences. By December 2021, two years into the pandemic, 67,000 genomes per day were being deposited into public viral genome data repositories such as GISAID, COG-UK and GenBank, which currently contain over 6 million SARS-CoV-2 genomes12,13,14 (Fig. 1a and Supplementary Table 1). The unprecedented volume of data collection for SARS-CoV-2 is evident by contrast with HIV genomic data collection: for HIV, which has consistently held the attention of public health officials and the general public since the 1980s, fewer than 16,000 full-length genome sequences have been collected by the biggest public HIV sequence database, at the Los Alamos National Laboratory in the United States, over the past 40 years15 (Fig. 1a).
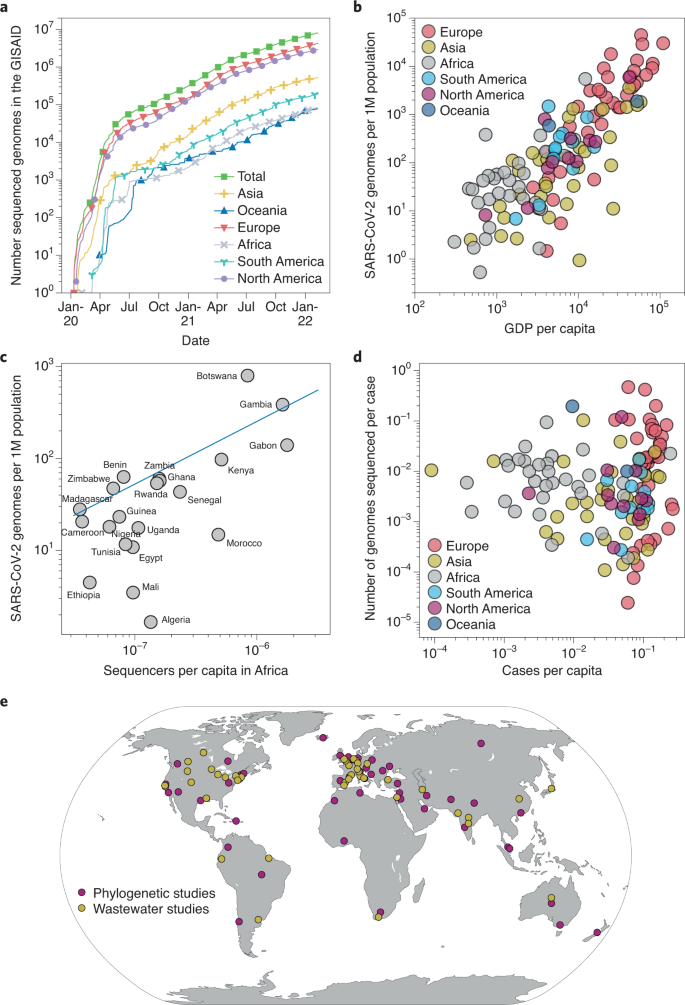
SARS-CoV-2 sequencing data collected all over the world and rapidly shared in online databases ultimately aided public health officials and governments in making better-informed decisions16. However, to fully explore the potential of such databases, a few issues still need to be resolved. Despite the unprecedented pace overall, inevitable delays caused by shortage of sequencing capacity, and in some regions political interference, led to problems in the logistical chain in these regions, including in sample collection, transporting and shipping samples17. Depending on the country and the strength of its public health infrastructure, the median time lag from collection to submission can differ greatly, ranging from one day to one year. Several factors influence the rate and scale of viral genomic sequencing across the globe. Countries with minimal sequencing capacity are likely to encounter outbreaks of higher severity, leading to blind spots of genomic surveillance that can facilitate the spread of new variants to other countries17. On average, high-income countries shared about 100 times more sequences per capita than low-income countries (Fig. 1b and Supplementary Fig. 2). However, some African countries with a low GDP per capita were able to sequence a comparable number of viral genomes to middle- and high-income countries18. This preparedness can be attributed to previous global initiatives to support African countries in mitigating outbreaks of other viruses that have enhanced sequencing capacities in the region. Africa provides a remarkable example of the necessity of international cooperation and of approaches that could be implemented in other parts of the world to improve pandemic response globally (Fig. 1c). In general, however, the number of shared coronavirus genomes per capita is correlated with the country’s GDP per capita (Fig. 1d).
Moving forward, several important data-sharing issues need to be addressed to facilitate open and rapid sharing of viral genome data. For example, it is important that scientists depositing sequencing data be able to trust that their rights will be respected by data users and that their authorship rights will not be violated19. The GISAID data access mechanism proved its ability to address these concerns and overcome obstacles to the international sharing of virus data, making GISAID the largest repository of influenza and SARS-CoV-2 genomic data16,20.
Bioinformatics methods cab accurately track SARS-CoV-2 genomic evolution
As SARS-CoV-2 spread through the world population over the first year of the pandemic, it gradually evolved into several viral lineages21,22,23,24. Statistical analysis of collected SARS-CoV-2 genomes showed that SARS-CoV-2 has a mutation rate of at least tenfold lower than that of seasonal influenza25. This lower mutation rate initially gave hope for efficient control of the pandemic through vaccination because the slower a virus mutates, the less chances it has to adapt to vaccines. However, given the large number of COVID-19 cases (>277 million and climbing, according to the WHO) and possibly because of SARS-CoV-2 recombination events, new variants continue to evolve, which are currently being classified as variants under investigation (VUIs), of interest (VOIs) and of concern (VOCs) according to their epidemiological, biological and/or immunological properties. Indeed, some variants acquired numerous mutations in a rapid fashion (variants Alpha and Omicron) and/or showed evidence of immune escape (variant Omicron). Notably, it was observed that immunodeficient individuals who experience unusually long periods of SARS-CoV-2 infection can provide a plausible environment for faster SARS-CoV-2 evolution because their immune systems allows viral immune escape26.
Before the COVID-19 pandemic, the public health community had had experience tracking and responding to genome evolution of viruses such as the influenza viruses that cause season flu. The Global Influenza Surveillance and Response System (GISRS) was established by the WHO for timely collection and genetic and antigenic characterization of these viruses27. Sharing of virus sequence data in the GISAID database along with the Nextstrain28 online phylogenetic tool are used for biannual selection of influenza A and B vaccine seed strains and to help understand viral genomic evolution and antigenic drift. GISAID and Nextstrain were both promptly adopted for collecting and analyzing SARS-CoV-2 genomic data, becoming the largest global system for tracking SARS-CoV-2 evolution and monitoring new variants.
The widespread application of sequencing technologies became possible because of extensive efforts by the scientific community to benchmark and standardize sequencing protocols and open-source bioinformatics workflows for accurate consensus genome assembly29. However, the use of proprietary next-generation sequencing solutions and software has been more commonplace in well-resourced national and state/province-level public health labs. The accessibility of tiled primer sequences (such as ARCTIC or midnight primer sets) and lower costs of Illumina and Oxford Nanopore sequencing, along with open-access bioinformatics workflows, supported sequencing in dozens of regional public health labs and academic institutions across the world. By 24 December 2021, 80.49% of available SARS-CoV-2 genomic data at GISAID had been generated by Illumina sequencers, 12.46% by Oxford Nanopore, 3.85% by Pacbio, 1.59% by IonTorrent, 1.29% by BGI, 0.31% by Sanger and 0.02% by Qiagen (Supplementary Fig. 1a). NCBI GenBank contains 91.04% genomic data sequenced by Illumina, 8.1% by Oxford Nanopore, 0.47% by IonTorrent, < 0.01% by PacBio and 0.38% unspecified (Supplementary Fig. 1b).
This democratization of viral sequencing methods has helped build pathogen sequencing capacity in low- to middle-income countries and has fostered insights into the genomic epidemiology of SARS-CoV-2, including the emergence and spread of variants, for example in Colombia (VOI Mu), Ukraine (VOC Delta), the Philippines (VOC Alpha), the UK (VOC Alpha, as it moved to the United States) and South Africa, where immune-evasive VOC Omicron was identified by genome sequencing30,31,32,33.
Bioinformatics methods enable tracking COVID-19 geographical spread in real time
As viruses evolve, tracking the appearance of new mutations and the locations where they were introduced can reveal geographical transmission routes. These routes help distinguish imported cases from those due to community transmission, aiding the identification of high-risk transmission routes that can be subject to enhanced public health control34. Comparative genomic analyses to study COVID-19 outbreak transmission dynamics have mostly been conducted using classic maximum-likelihood (ML) phylogenetic methods35. Unfortunately, ML methods are not scalable enough to handle the large volumes of SARS-CoV-2 genomic data available. For ML, therefore, it is often necessary to reduce sample size and consider only a fraction of the data in order to conduct the analysis, which can potentially compromise the accuracy of the results. Alternatively, more scalable approximate maximum-parsimony methods (MP) can be used for phylogeny reconstruction from dense SARS-CoV-2 data36. Indeed, it has been shown theoretically that with dense enough sampling, MP produces an ML tree under certain ML models37,38,39. Another approach has been to use network-based methods, which are significantly faster but theoretically less accurate than phylogeny-based methods40,41,42.
The public availability of diverse SARS-CoV-2 genome sequences from around the world has facilitated the efficient and accurate tracking of local and global SARS-CoV-2 transmission routes43,44,45 (Supplementary Fig. 3). Phylogenetics methods (Supplementary Table 2) revealed that SARS-CoV-2 was introduced into Europe from China and into the United States from China and Europe34,46,47,48 and have also been used to track domestic transmission chains and differentiate them from international ones. In the United States, for example, studies showed that SARS-CoV-2 was likely introduced into Connecticut via a domestic transmission route, and the most successful viral introductions in Arizona were also likely via domestic travel34,49. The New York City area experienced multiple introductions of SARS-CoV-2, primarily from Europe50. Similarly, phylogenetic analysis suggested that SARS-CoV-2 was likely introduced into France from several countries, including China, Italy, the United Arab Emirates, Egypt and Madagascar51 (Fig. 1e and Supplementary Table 2).
Differences in sampling across geographical locations and over time represent a considerable challenge to the accurate reconstruction of spatial transmission patterns. However, additional data, such as travel information and epidemiological estimates, may help mitigate difficulties due to non-uniform sampling across geographical locations and time and may contribute to a more complete picture of viral spread. This has been illustrated by a study of SARS-CoV-2 importation and establishment in the UK52. Large-scale genomic data resulted in estimates of the number and timing of introductions events, but combining these data with epidemiological and travel data made it possible to identify the spatiotemporal origins of these introductions. Such additional data sources are also increasingly being integrated into phylodynamic inferences. For example, a study of the contribution of persistence versus new introductions to the second COVID-19 wave in Europe made use of Google mobility data to inform the phylogeographic component of the genomic reconstruction53. The individual travel history of sampled individuals can also be formally incorporated into such analyses54.
Additionally, phylogenetics can be used to monitor the effectiveness of global travel restrictions and lockdowns. For example, it was shown that the risk of domestic transmission of SARS-CoV-2 in Connecticut already exceeded that of international introduction at the time federal travel restrictions were imposed, highlighting the critical need for local surveillance34. Similarly, in Brazil, three clades of European origin were established before the initiation of travel bans and lockdowns55. In the UK, lineages introduced before national lockdown were shown to be larger and more dispersed, and lineage importation and regional lineage diversity declined after lockdown52. Phylogenetics showed that several international introductions of SARS-CoV-2 likely occurred in Morocco as a result of violations of imposed lockdowns involving sea trade56. In Australia, lockdown effectiveness was validated using SARS-CoV-2 genomic data coupled with agent-based modeling, a computation tool to simulate the interactions of autonomous agents such as individuals57. Phylogenetic modeling of over 11,000 SARS-CoV-2 genomes collected in Switzerland throughout 2020 enabled estimation of the effects of different public health measures, including lockdown, border closure and test–trace–isolate efforts58. Similarly, comparative phylodynamics analysis of SARS-CoV-2 transmission dynamics in the neighboring Eastern European countries of Belarus and Ukraine, which followed highly different COVID-19 containment policies, allowed an assessment of the effectiveness of public health intervention measures in this region, and highlighted the roles of regional political and social factors in virus spread59.
Genomics methods enable wastewater-based monitoring of SARS-CoV-2 epidemiology
The presence of trace viral genomic material in wastewater has been successfully exploited to track antibiotic use60 and tobacco consumption61 and for the monitoring of several respiratory and enteric viruses, including poliovirus62. Although COVID-19 is primarily associated with respiratory symptoms, SARS-CoV-2 is regularly shed in the feces of infected individuals63. As of December 2021, wastewater-based surveillance to track SARS-CoV-2 viral infection dynamics64 had been implemented in many countries around the world (Fig. 1e).
Wastewater-based epidemiology has been shown to provide more balanced estimates of viral prevalence rates in a population than clinical testing alone due to inherent limitations in testing resources and/or testing uptake rates, especially in underserved communities. Combining clinical diagnostics with wastewater-based surveillance can provide a more comprehensive community-level profile of both symptomatic and asymptomatic cases, enabling identification of hospital capacity needs65,66,67,68,69,70,71,72. Another important advantage of wastewater monitoring is the ability to detect early-stage outbreaks before they become widespread62,73,74,75,76. Although tracking of SARS-CoV-2 viral RNA via quantitative PCR (qPCR)-based methods can reveal temporal changes of virus prevalence in a given population, it cannot provide underlying epidemiological information to identify transmission or genomic details of emerging variants. Tracking viral genomic sequences from wastewater significantly improves community prevalence estimates and also provides detection of emerging variants. Tracking SARS-CoV-2 viral genomic sequences from wastewater using a targeted tiled amplicon-based sequencing approach would significantly ameliorate community prevalence estimates and also detect emerging variants77.
Wastewater genomic epidemiology can also act as a surrogate for elucidating strain geospatial distributions, helping identify outbreak clusters and track prevailing and newly emerging variants, and covering even areas with insufficient clinical testing rates. However, the highly variable nature of wastewater, low viral loads, fragmented RNA and the presence of multiple genotypes in a single sample makes it challenging to obtain good-quality genome sequences and discern lineages with a high degree of accuracy78.
The commonly used tools used for discerning viral lineages in clinical samples, such as pangolin3 and UShER79, cannot deconvolute the multiple lineages that are commonly observed in a single wastewater sample and at best detect the most dominant one. As existing lineage-calling methods require a single consensus sequence to perform assignment, they are ill-equipped to capture the diversity present in mixed viral samples. Hence, tools to robustly identify the multiple lineages and their relative proportions present in wastewater are critical in understanding and interpreting the underlying sequence data obtained from these samples. For example, a depth-weighted demixing algorithm, Freyja80, employs a ‘barcode’ library of lineage-defining mutations to represent each viral variant and can be used to recover relative abundances of different lineages within samples. This approach enabled the early detection of emerging VOCs in wastewater up to 14 days before their first clinical detection and also identified multiple instances of cryptic transmission not observed via clinical genomic surveillance81. Similar algorithms for mutation calling, haplotype reconstruction and population characterization in viral specimens can also be used to deconvolute the mixture of variants present in a wastewater sample82,83. By searching for signature mutations co-occurring on the same amplicon, variant B.1.1.7 was detected in wastewater eight days before the first patient sample tested positive for the variant84. Similarly, RNA transcript quantification methods, such as Kallisto, can be used to estimate the relative abundance of SARS-CoV-2 variants in wastewater85. Both digital PCR-based and sequencing-based estimates of variant abundance in wastewater have been used to derive the fitness advantage of a recently introduced variant, an important epidemiological parameter for assessing the expected transmissibility and spread of such a variant84,86.
Alternatively, viral genomes in wastewater can be sequenced via next-generation sequencing approaches after enriching for a wider array of RNA viruses present in a sample through a hybrid probe-capture approach. This approach allows characterization of the prevalent SARS-CoV-2 genomic variants in a defined local region and the dynamics of other pathogenic viruses present in the sample87,88,89. Shotgun metagenomic and metatranscriptomic sequencing (i.e., community-based sequencing approaches) can provide a comprehensive snapshot of the viral community ecology and thereby aid the tracking of viruses of clinical significance in a community.
As SARS-CoV-2 transitions to become an endemic pathogen, wastewater genomic sequencing offers a scalable, less expensive, long-term passive surveillance tool to track emerging variants in the population. A global metagenomics approach has been suggested to detect, collect and store samples in preparation for future pandemics90,91. Resources such as GISAID, GenomeTrakr92,93 and the US National Wastewater Surveillance System (CDC-NWSS)94 could facilitate the above efforts.
Outlook
The unprecedented volume of available SARS-CoV-2 genomic data coupled with available bioinformatics tools accelerated the prompt and effective characterization of SARS-CoV-2 genomes and provided tools enabling epidemiologists and public health officials to more effectively respond to the COVID-19 pandemic. Numerous independent efforts across the globe used bioinformatics methods, thereby demonstrating the utility of genomics-based approaches and creating a solid foundation for the response to COVID-19 and future pandemics. This was achieved by the standardization of methodology, protocol and data sharing, and applications of SARS-CoV-2 genomic data in epidemiological investigations.
Genome-based surveillance has been shown to be beneficial in addressing COVID-19. However, the unprecedented volume of sequencing data, currently six million complete SARS-CoV-2 genome sequences in databases, pose a challenge to the current systems of data storage, processing and bioinformatics analysis16,19,95. Owing to various technological burdens, such systems were still in the early stages of development when SARS-CoV-19 emerged in December 2019. COVID-19 has led to the mobilization of financial, scientific and developmental resources in record time, with numerous global surveillance systems providing resources for outbreak response using SARS-CoV-2 genome analysis (Table 1). A notable example is the timely deployment of GISAID and Nextstrain to address the COVID-19 response. This technology has played a leading role in centralizing efforts to collect and analyze SARS-CoV-2 genomic data.Table 1 Online services with SARS-CoV-2 genome resources and analytics
Emerging VOCs, VOIs and VUIs are likely to continue shaping the course of the COVID-19 pandemic. Global genomics-based surveillance for new variants, in our view, will continue to play a leading role, with information on all SARS-CoV-2 lineages being collected and made available online for the rapid evaluation of their impact on transmission, virulence and vaccine escape96,97. We believe that targeted genomic surveillance of SARS-CoV-2 in immunocompromised patients can provide useful insights into the mechanisms of appearance of newly emerging VOCs. This can be done by applying bioinformatics tools for intra-host population analysis similar to those that are already available for other RNA viruses, such as HCV and HIV82,98,99,100,101.
Efficient early detection and tracking of potentially dangerous variants requires real-time data from all countries102. The European Commission, for example, recommended achieving a capacity to sequence at least 5% of positive test results, which can be a good global standard. Yet, many underdeveloped countries face insurmountable logistic, technological and financial barriers to operating sequencing centers to accommodate this scale of testing, suggesting that developed countries should share responsibility for global surveilance103. Following the example of many African countries, countries in other regions that are currently lacking in viral genomic sequencing capability could establish additional sequencing centers. In regions where that is not practical, a logistically efficient system to obtain samples and deliver them to sequencing centers in other countries might be an appealing alternative.
In our view, there are three potential benefits of a standard genome epidemiological sequencing system. The immediate benefit is that this improve the timeliness and accuracy with which emerging VOIs and VOCs can be tracked. A longer-term goal is an improved ability to learn about the evolutionary pressures driving the emergence of novel, potentially dangerous variants. Presently, VOCs are declared based on their increased transmissibility or virulence, or decreased effectiveness of public health and social measures, available diagnostics, vaccines and therapeutics. Learning more about the evolutionary dynamics of emergent strains may lead to predictions of VOIs based on genomic sequence alone, further improving response times. Finally, a truly global system of pathogen genome sequencing and analysis is likely to improve our ability to combat future pandemics.
Global coordination of genomic data surveys will also allow wider application of wastewater-based or environmental-based virus surveillance104. Currently, wastewater-based monitoring lacks the granularity of clinical diagnostic testing and cannot discern a particular area of an outbreak when the wastewater treatment plant serves a large population. Sampling at a higher spatial resolution within the sewer system, or even at a building-level scale, could potentially provide early indications of viral outbreaks and help monitor their progression105.