Mechanisms Underlying Disease Severity and Progression
Authors: Mary Kathryn Bohn,1,2, Alexandra Hall,1 Lusia Sepiashvili,1,2, Benjamin Jung,1,2 Shannon, Steele,1 and Khosrow Adeli1,2,3
The global epidemiology of coronavirus disease 2019 (COVID-19) suggests a wide spectrum of clinical severity, ranging from asymptomatic to fatal. Although the clinical and laboratory characteristics of COVID-19 patients have been well characterized, the pathophysiological mechanisms underlying disease severity and progression remain unclear. This review highlights key mechanisms that have been proposed to contribute to COVID-19 progression from viral entry to multisystem organ failure, as well as the central role of the immune response in successful viral clearance or progression to death.
Introduction
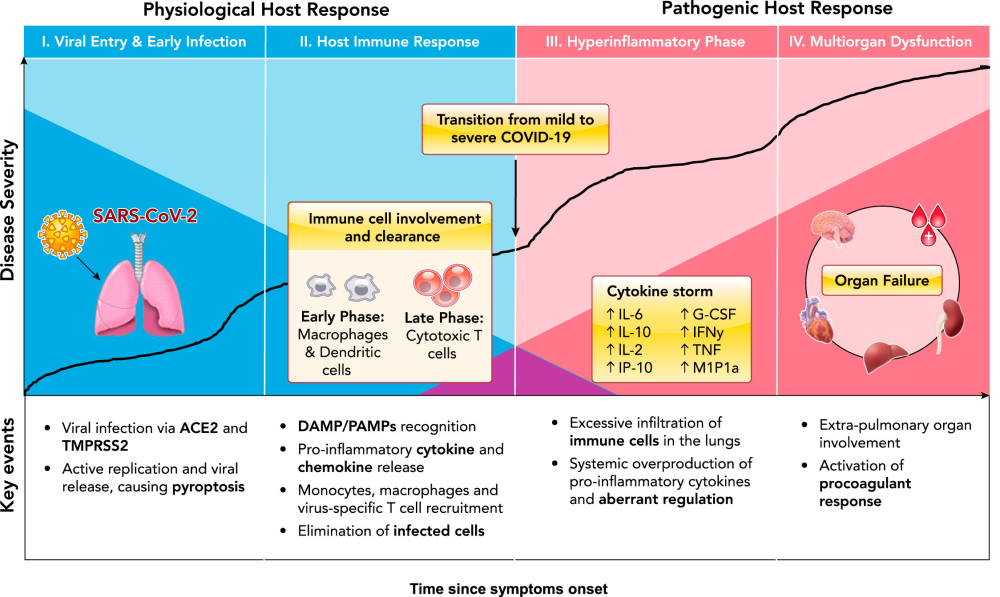
Coronavirus disease 2019 (COVID-19) is caused by a novel beta-coronavirus known as Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). As of June 15, 2020, the number of global confirmed cases has surpassed 8 million, with over 400,000 reported mortalities. The unparalleled pathogenicity and global impact of this pandemic has rapidly engaged the scientific community in urgently needed research. Preliminary reports from the Chinese Center for Disease Control and Prevention have estimated that the large majority of confirmed SARS-CoV-2 cases are mild (81%), with ~14% progressing to severe pneumonia and 5% developing acute respiratory distress syndrome (ARDS), sepsis, and/or multisystem organ failure (MOF) (144). Although more data is urgently needed to elucidate the global epidemiology of COVID-19 (80), a wide spectrum of clinical severity is evident, with most patients able to mount a sufficient and appropriate immune response, ultimately leading to viral clearance and case resolution. However, a significant subset of patients present with severe clinical manifestations, requiring life-supporting treatment (51). The pathophysiological mechanisms behind key events in the progression from mild to severe disease remain unclear, warranting further investigation to inform therapeutic decisions. Here, we review the current literature and summarize key proposed mechanisms of COVID-19 pathophysiological progression (FIGURE 1). Key Pathophysiological Mechanisms: Our Current Understanding Viral Invasion The first step in COVID-19 pathogenesis is viral invasion via its target host cell receptors. SARSCoV-2 viral entry has been described in detail elsewhere (138). In brief, SARS-CoV-2 consists of four main structural glycoproteins: spike (S), membrane (M), envelope (E), and nucleocapsid (N). The M, E, and N proteins are critical for viral particle assembly and release, whereas the S protein is responsible for viral binding and entry into host cells (33, 76, 89, 143, 148). Similar to SARS-CoV, several researchers have identified human angiotensin converting enzyme 2 (ACE2) as an entry receptor for SARS-CoV-2 (75, 99, 148, 156). SARSCoV-2 is mostly transmissible through large respiratory droplets, directly infecting cells of the upper and lower respiratory tract, especially nasal ciliated and alveolar epithelial cells (161). In addition to the lungs, ACE2 is also expressed in various other human tissues, such as the small intestine, kidneys, heart, thyroid, testis, and adipose tissue, indicating the virus may directly infect cells of other organ systems when viremia is present (77). Interestingly, although the S proteins of SARS-CoV-2 and SARSCoV share 72% homology in amino acid sequences, SARS-CoV-2 has been reported to have a higher affinity for the ACE2 receptor (18, 21, 143). Following host cell binding, viral and cell membranes fuse, enabling the virus to enter into the cell (89). For many coronaviruses, including SARS-CoV, host cell binding alone is insufficient to facilitate membrane fusion, requiring S-protein priming or cleavage by host cell proteases or transmembrane serine proteases (9, 10, 90, 94, 108). Indeed, Hoffman and colleagues demonstrated that S-protein priming by transmembrane serine protease 2 (TMPRSS2), which may be substituted by cathepsin B/L, is required to facilitate SARS-CoV-2 entry into host cells (58). In addition, unlike other coronaviruses, SARS-CoV-2 has been reported to possess a furin-like cleavage site in the S-protein domain, located between the S1 and S2 subunits (31, 138). Furin-like proteases are ubiquitously expressed, albeit at low levels, indicating that S-protein priming at this cleavage site may contribute to the widened cell tropism and enhanced transmissibility of SARS-CoV-2 (123). However, whether furin-like protease-mediated cleavage is required for SARS-CoV-2 host entry has yet to be determined. Blocking or inhibiting these processing enzymes may serve as a potential antiviral target (130). Interestingly, SARS-CoV-2 has developed a unique S1/S2 cleavage site in its S protein, characterized by a four-amino acid insertion, which seems to be absent in all other coronaviruses (4). This molecular mimicry has been identified as an efficient evolutionary adaptation that some viruses have acquired for exploiting the host cellular machinery. Once the nucleocapsid is deposited into the cytoplasm of the host cell, the RNA genome is replicated and translated into structural and accessory proteins. Vesicles containing the newly formed viral particles are then transported to and fuse with the plasma membrane, releasing them to infect other host cells in the same fashion (33, 89, 105). Although much progress has been made in our understanding of the mechanisms underlying SARS-CoV-2 invasion, additional research is needed to delineate exactly how cleavage of the S proteins by TMPRSS2 confers viral particle entry as well as how S-protein cleavage by membrane proteases contributes to viral penetration.
For More Information: https://journals.physiology.org/doi/pdf/10.1152/physiol.00019.2020