Authors: Flor M. Munoz,a,⁎Jakob P. Cramer,bCornelia L. Dekker,cMatthew Z. Dudley,dBarney S. Graham,eMarc Gurwith,fBarbara Law,gStanley Perlman,hFernando P. Polack,iJonathan M. Spergel,jEva Van Braeckel,kBrian J. Ward,lArnaud M. Didierlaurent,mPaul Henri Lambert,m and for the Brighton Collaboration Vaccine-associated Enhanced Disease Working Group
Abstract
This is a Brighton Collaboration Case Definition of the term “Vaccine Associated Enhanced Disease” to be utilized in the evaluation of adverse events following immunization. The Case Definition was developed by a group of experts convened by the Coalition for Epidemic Preparedness Innovations (CEPI) in the context of active development of vaccines for SARS-CoV-2 vaccines and other emerging pathogens. The case definition format of the Brighton Collaboration was followed to develop a consensus definition and defined levels of certainty, after an exhaustive review of the literature and expert consultation. The document underwent peer review by the Brighton Collaboration Network and by selected Expert Reviewers prior to submission.Keywords: Adverse event, Immunization, Guidelines, Case definition, Vaccine, Enhanced disease, Respiratory, Systemic diseaseGo to:
1. Preamble
Vaccine-associated enhanced diseases (VAED) are modified presentations of clinical infections affecting individuals exposed to a wild-type pathogen after having received a prior vaccination for the same pathogen [1]. Vaccine-associated enhanced respiratory (VAERD) disease refers to disease with predominant involvement of the lower respiratory tract. Classic examples of VAED are atypical measles and enhanced respiratory syncytial virus (RSV) occurring after administration of inactivated vaccine for these pathogens. In this situation, severe disease has been documented resulting from infection in individuals primed with non-protective immune responses against the respective wild-type viruses [2], [3], [4], [5], [6]. Given that these enhanced responses are triggered by failed attempts to control the infecting virus, VAED typically presents with symptoms related to the target organ of the infection pathogen. In order to recognize vaccine associated disease enhancement, it is therefore necessary to have a clear understanding of the clinical presentation and usual course of the natural disease.
Disease enhancement independent of vaccine priming has also been described for pathogens causing sequential infections with different cross-reactive but not cross-protective serotypes, including dengue and pandemic influenza [7], [8], [9], [10], [11], [12].
In late 2019, a novel severe respiratory illness emerged in Wuhan, China [13]. The causative agent, Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), was promptly identified, and determined to be closely related to SARS and the Middle East Respiratory Syndrome (MERS) coronaviruses, which had caused geographically localized outbreaks in 2002–2004 and from 2012 onwards, respectively. SARS-CoV-2 progressed to a global pandemic with substantial consequences due to its high infectivity and transmissibility, and its ability to cause both a severe respiratory illness, and a systemic disease with fatal consequences for vulnerable populations. The natural history of coronavirus infectious disease caused by SARS-CoV-2 (COVID-19), is yet to be fully described. However, a case fatality rate that ranges from 0.5% to nearly 20% depending on age and other risk factors, and the understanding that SARS-CoV-2 is now a well-adapted human pathogen that will continue to cause disease in susceptible populations, makes the development of an effective vaccine a global priority.
The potential for vaccination against SARS-CoV-2 to be associated with disease enhancement is of theoretical concern, given similar observations with other respiratory viruses in general, and in animal models of highly pathogenic coronaviruses in particular [14]. Importantly, VAED has not been seen following SARS or MERS vaccines given to humans, albeit the number of people who received these experimental vaccines remains very small. At this time, the pathogenesis, host responses and immunity to SARS-CoV-2 are still being evaluated and are not fully understood. SARS-CoV-2 infection is associated with a spectrum of disease that varies from asymptomatic infection to severe lung disease with acute respiratory distress syndrome (ARDS) and a fatal multiorgan disease with inflammatory, cardiovascular, hematologic and coagulation dysregulation [15], [16], [17]. Post-infectious, possibly immune-mediated systemic disease has also been described, particularly the multisystemic inflammatory syndrome in children (MIS-C) and adults (MIS-A) of unclear pathogenesis at this time [18], [19], [20], [21].
Given the broad spectrum of disease associated with SARS-CoV-2, clinical assessment of both systemic VAED and lung-specific VAERD will be challenging during the pre-licensure evaluation of candidate vaccines and after the implementation of widespread vaccination for COVID-19. The broad spectrum of natural disease manifestations in different populations and age groups makes it very difficult, if not impossible, to determine how severe COVID-19 infection would have been in the absence of vaccination in the individual case. Someone who might have been completely asymptomatic without prior vaccination but who develops mild respiratory symptoms in association with prior vaccination could logically be considered a case of VAERD. However, this end of the spectrum of possible VAERD would have very little clinical significance for this individual person. At the population level however, even a small shift in the spectrum of disease towards greater severity could have major clinical and societal impact. Furthermore, given that severe illness is more feasible to detect and characterize, the case definitions discussed herein focus on the more severe presentations of VAED/VAERD.
There is no uniformly accepted definition of VAED or VAERD. Frequently used related terms include “vaccine-mediated enhanced disease (VMED)”, “enhanced respiratory disease (ERD)”, “vaccine-induced enhancement of infection”, “disease enhancement”, “immune enhancement”, and “antibody-dependent enhancement (ADE)”. This is potentially confusing as the mechanisms for disease enhancement likely vary, and data comparability across trials or surveillance systems can be problematic when the systems do not utilize a consistent case definition and do not collect comparable data. However, the assessment of this potential adverse event following immunization is particularly important for SARS-CoV-2, given the urgent global need for safe and effective vaccines. While this case definition was developed for the identification of potential cases of VAED/VAERD in the context of SARS-CoV-2 vaccine development, it is not exclusive for COVID-19 vaccines and may be applied in the evaluation of possible VAED/VAERD after any vaccine.
1.1. Methods for the development of the case definition
The Brighton Collaboration VAED working group was formed in March 2020 and included members with expertise in basic science, virology, animal models, immunology, vaccinology, vaccine safety, clinical care, clinical research, public health, regulatory sciences and ethics.
To guide the decision-making for the case definition and guidelines, a series of literature searches were performed using PubMed. The search terms and results of the searches are described in detail in [99], [100], [101], [102], [103], [104], [105], [106], [107], [108], [109], [110], [111], [112], [113], [114], [115], [116], [117], [118], [119], [120], [121], [122], [123], [124], [125], [126], [127], [128].
To address the current state of knowledge and knowledge gaps for the assessment of VAED in the context of the assessment of vaccines for SARS-CoV-2, a Consensus Conference of Experts, including the authors of this case definition, was convened on March 12–13, 2020. The topics of discussion and conclusions of this meeting are published [22]. The group of experts in this Consensus Meeting concluded that the demonstration of some disease enhancement with any candidate vaccine after viral challenge in animal models should not necessarily represent a “no-go” signal for deciding whether to progress into early trials in clinical development of a COVID-19 vaccine. However, continuous monitoring of this risk during clinical trials in an epidemic context will be needed. Each observed effect should be discussed by the vaccine developers with the respective regulatory agencies who will ultimately define the actual requirements for clinical studies.
The working group determined that describing the known pathophysiologic pathways leading to VAED/VAERD was necessary to highlight the different potential mechanisms of disease, because an established clinical, laboratory or histopathologic definition of VAED or VAERD is not available. The case definition focuses on the identification of possible and probable cases of VAED/VAERD after any vaccination based on clinical presentation and frequency of clinical outcomes of concern, and provides suggested clinical and laboratory evaluation tools. However, while describing options for evaluation of possible cases when planning clinical trials or safety surveillance, a specific biomarker or histopathologic finding of VAED does not exist, and therefore, the case definition is not prescriptive regarding the specific tests to perform, nor when and who should be conducting or interpreting such testing. Furthermore, with the current state of knowledge, the “gold standard” evaluation to diagnose a definitive case of VAED/VAERD may be pathogen- and vaccine-specific and cannot be defined until more clinical and research data become available, in consultation with experts.
1.2. Defining pathophysiologic pathways leading to VAED/VAERD
Previous experiences of specific vaccine enhanced diseases serve as examples of how various pathophysiologic mechanisms can lead to VAED or VAERD. The mechanisms are outlined below.
1.2.1. Immune complex mediated enhanced disease (RSV, measles, pandemic influenza)
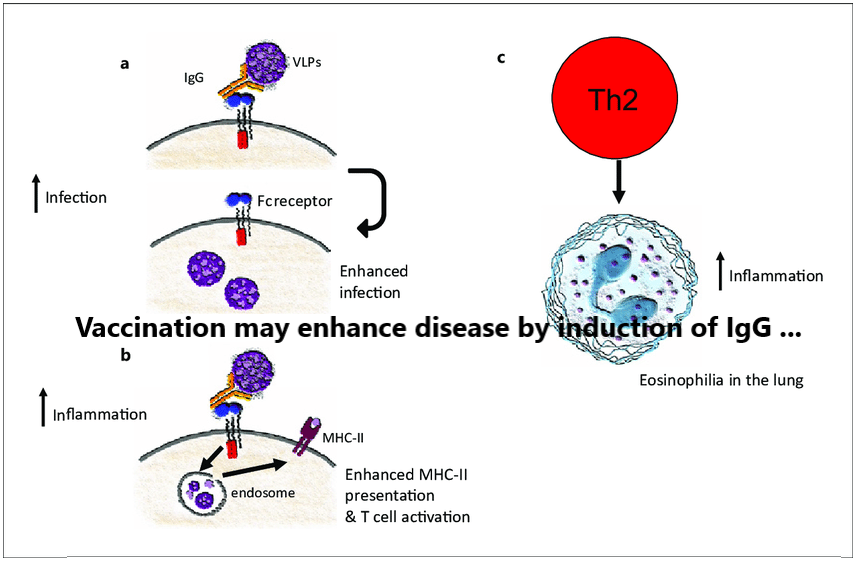
Shortly after the successful inactivation of polioviruses with formaldehyde, and the success of that vaccine for the control of epidemic polio, other pediatric pathogens were targeted for vaccine development using similar methods. In the mid-1960s, a formalin-inactivated vaccine against RSV was administered to infants and young children in four studies in the United States [23], [24], [25], [26]. Children were subsequently exposed to wild-type virus in the community, and those immunized children who were seronegative for RSV before vaccination experienced an enhanced and atypical presentation of RSV disease, with fever, wheezing, and bronchopneumonia. These children were more frequently hospitalized and two children vaccinated in infancy, died as a consequence of the RSV infection [23]. In contrast, enhanced respiratory disease (ERD) was not observed in infants who were seropositive for the virus at the time of administration of the formalin-inactivated RSV vaccine [23].
The potential reason for the enhanced disease associated with the formalin-inactivated vaccine was that a non-protective antibody response of low affinity for the RSV fusion (F) protective antigen was generated [27], [28], [29]. This low-avidity, non-protective response elicited by formalin-inactivated vaccines has been linked to deficient Toll-like receptor (TLR) activation at the time of immunization [27], [28], [29], [30], [31]. Subsequent RSV infection leads to immune complex formation and complement activation with pulmonary injury, exacerbation of bronchopneumonia with a Th2 biased CD4 T-cell response (a distinctive phenotype of the disease) [32], [33], [34], [35], [36] and abundant mucus production [37], [38]. Another potential contributing factor to non-protective antibody responses elicited by formalin-inactivated vaccine may have been the administration of RSV F in its post-fusion conformation, which is less stable and results in antibodies with lower neutralizing capacity than the pre-fusion conformation [30].
Interestingly, affinity maturation during an earlier exposure to RSV explains why children who were seropositive for the virus before inoculation never developed ERD. Preexisting acquisition of high-avidity antibodies against wild-type RSV likely outcompeted low-affinity B cell clones elicited by the formalin-inactivated RSV vaccine thus eliminating the low affinity non-protective B cells. This same paradigm explains why no children experienced ERD twice. After experiencing RSV disease enhancement post-wild-type infection, new antibodies with high affinity for the virus in these individuals established a healthy response against subsequent reinfections.
A formalin-inactivated vaccine against measles virus was licensed in the United States in 1963 simultaneously with the first live-attenuated measles vaccine [39], [40]. Although most people were initially protected by the formalin-inactivated vaccine, the relatively low-avidity antibodies elicited by this vaccine failed to protect at lower titers and led to a severe form of illness known as atypical measles, in immunized individuals exposed to wild-type virus [41]. Children with atypical measles presented with high fever, a petechial rash in the extremities, and bibasilar pneumonia [42]. In this case, low-avidity antibodies elicited by the vaccine failed to neutralize virus that bound to the CD150 high-affinity receptor in exposed individuals, and promoted immune complex-mediated illness at sites of measles virus infection, mainly the skin and lungs [43], [44], [45], [46]. Importantly, atypical measles occurred many years after exposure to the formalin-inactivated vaccine.
After observing the first cases of atypical measles, formalin-inactivated vaccine recipients not yet exposed to wild-type measles were inoculated with the licensed live-attenuated vaccine, with the intent of generating protective antibodies to prevent further atypical cases. These individuals developed an erythematous nodule at the subcutaneous injection site, characterized histopathologically by measles virus-specific immune complexes [47], [48]. Some failed to mount a corrective immune response following the live-attenuated vaccination, presumably due to neutralization of the vaccine-strain virus by pre-existing antibodies. In most however, the live attenuated vaccine successfully induced a high affinity IgG response that could outcompete the potentially pathogenic antibodies and provide long-term protection.
1.2.2. Cellular immunity in enhanced respiratory disease and atypical measles.
Atypical measles and ERD are also characterized by a Th2 polarization of their immune response. Although mice are not an ideal small animal model for either RSV or measles virus infection, an early evaluation of ERD pathogenesis by Graham et al., showed increased production of interleukin 4 (IL-4) in lungs of affected BALB/c mice [35]. Subsequent depletion of CD4+ T lymphocytes and co-depletion of IL-4 and IL-10 ameliorated ERD lung pathology, suggesting that the disease was due, at least in part, to an exacerbated Th2 response [29], [36]. These findings were expanded by reports of high levels of IL-5 and IL-13, increased numbers of eosinophils, and CD4+ T lymphocytes in mice with ERD [32], [33]. In recent years, a critical role for Th2 bias has been described for enhanced disease components, including airway hyperreactivity and mucus hypersecretion [32], [33]. Formaldehyde inactivation of RSV may also have contributed to Th2 polarization during ERD by generating carbonyl groups on viral antigens [48].
In addition, other T lymphocyte populations may have contributed to ERD pathogenesis and its phenotype. Marked suppression of T regulatory cells (T-reg) may have exacerbated the Th2 bias in formalin-inactivated RSV vaccine recipients; [49] absence of RSV-specific cytotoxic T lymphocytes response after immunization was permissive for viral replication in the lungs and contributed to a Th2 bias in the anamnestic CD4+ T lymphocyte response during wild-type infection; [50], [51] and eosinophils, though probably not a critical factor in disease pathogenesis [52], may be a useful biomarker of undesirable responses in animal models of disease.
1.2.3. Antibody-mediated enhanced disease (Dengue)
Dengue viruses (DENV) belong to the genus Flavivirus, with four serologically and genetically distinct serotypes, which differ by 30–35% amino acid identity. The symptoms of dengue infections range from asymptomatic in about two-thirds to mild flu-like symptoms to dengue fever. Dengue hemorrhagic fever/dengue shock syndrome is the most severe form of dengue disease and is characterized by vascular leakage, hemorrhagic manifestations, thrombocytopenia, and hypotensive shock. People exposed to their first DENV infection develop memory B cells and long-lived plasma cells that produce antibodies that can either be cross-reactive or specific to the serotype of infection. Secondary DENV infections induce cross-neutralizing antibodies and protective immunity. However, priming with one DENV serotype can sometimes increase the risk of severe dengue upon secondary infection with a different DENV serotype. The mechanism for increased disease severity is thought to be associated with antibody-dependent enhancement (ADE) [53], [54]. It has been postulated that low levels of antibodies or non-neutralizing antibodies induced by a previous DENV infection bind to the new serotype of DENV and facilitate viral entry into Fcγ receptor (FcγR)-bearing cells, leading to higher viremia and immune activation. Strong evidence for this ADE mechanism after natural infection comes from a pediatric cohort study in Nicaragua, where an increased risk for viremia and severe disease (7.64-fold higher, 95% confidence interval (CI): 2.19–18.28) was observed in children with preexisting DENV- antibody levels of 1:21–1:80 compared to DENV-naïve or those with high (>1:1280) antibody titers [55].
There is concern that dengue vaccines can induce similar ADE. If a vaccine produces antibodies with poor neutralizing activity that bind heterotypic virions without achieving neutralization, the opsonized viral particle may have an increased ability to infect Fcγ-R-bearing cells (i.e., facilitated entry). If a vaccine does not induce enough neutralizing antibodies against one or more serotypes, VAED may develop upon exposure to these serotypes. In a phase 3 clinical trial, children who received vaccine (Dengvaxia™: Sanofi Pasteur) had an increased risk of hospitalization due to dengue compared to the placebo group in year 3 after vaccination [9], [56]. While the mechanism of ADE is not proven, prior exposure to dengue (positive serostatus) is thought to play a critical role, and those that are dengue-naïve seem to have higher risk of severe disease after vaccination when subsequently infected with another serotype of dengue virus [57]. Nevertheless, the data for ADE are controversial as most of the evidence is anecdotal or based on animal models.
In in vitro and animal models, a peak enhancement titer (i.e., a specific concentration of antibodies that most efficiently enhances DENV infection) has been observed. By contrast, higher antibody concentrations effectively neutralize virions, whereas lower concentrations can enhance infection. However, neutralizing assays vary from laboratory to laboratory and standardization of such bioassays across laboratories can be challenging. ADE is also postulated for other arboviruses including Zika and Japanese Encephalitis Virus [58], [59], [60].
1.2.4. Cytokine activation/storm and enhanced disease (SARS, MERS, SARS-CoV-2)
Exuberant cytokine activation is considered an important component in severe disease caused by SARS-CoV, MERS-CoV or SARS-CoV-2. The precise mechanism of this immunopathologic response still remains unclear, since active cytokine/chemokine production may be an appropriate response to uncontrolled virus replication as opposed to a truly excessive response. In any case, prolonged cytokine responses in patients with SARS, characterized by expression of mainly INF-gamma [61], [62], are correlated with worse outcomes. In these patients, lymphopenia was commonly observed, which likely reflected effects of elevated levels of cytokines and endogenous corticosteroids. In one study, prolonged levels of type I interferon (INF) and other cytokines were observed in SARS patients who did poorly, while these levels were generally lower in patients who had better clinical responses, coincident with the development of protective antibody responses [63]. However, in other studies, antibody responses were higher in patients with worse outcomes, raising the possibility that the antibody response actually contributed to more severe disease. Macrophages are considered an important source of pro-inflammatory cytokines, but these cells are not productively infected with SARS-CoV [64], [65]. Both macrophages and dendritic cells are abortively infected with SARS-CoV. While the cells do not support productive infection, in acute respiratory distress syndrome (ARDS) they were shown to produce pro-inflammatory cytokines such as TNF, IL-1beta, IL-6, IL-8, CCL2 and CCL7 [66], [67], [68]. Additional insight into the role of excessive cytokine activation in SARS comes from mouse studies. Mice infected with mouse-adapted SARS-CoV develop a lethal pneumonia, characterized by rapid virus replication and peak virus titers within 16–24 h. However, 100% of mice survive if type I IFN signaling is blocked either by genetic deletion or treatment with antibody that blocks INF signaling [69].
Similar mechanisms appear to occur in MERS patients, although less is known because there have been only approximately 2500 cases since MERS was first identified in 2012. As in SARS, MERS patients tend to have (delayed) elevated levels of pro-inflammatory cytokines such as IL-1beta, IL-6 and IL-8 [70]. MERS-CoV, unlike SARS-CoV, actively inhibits the induction of an early IFN-I response, allowing for enhanced virus replication [71]. Also unlike SARS-CoV, MERS-CoV is sensitive to IFN-I therapy [71]. MERS-CoV was shown to productively infect macrophages and dendritic cells, with delayed induction of IFN-I and other cytokines [72]. Thus both SARS-CoV and MERS-CoV induce the expression of pro-inflammatory molecules, even though their ability to replicate in myeloid cells differs substantially.
Although SARS-CoV-2 has been in human populations for only a few months, several studies have suggested that excessive cytokine activation contributes to pathogenesis. Molecules, such as IL-1, IL-6, TNF, IL-8 are upregulated in patients with more severe disease, raising the possibility that some of them may contribute to poor outcomes [73]. IL blocking antibodies are being used clinically in both controlled and uncontrolled clinical trials.
ADE has only been convincingly demonstrated in coronavirus infections in cats that were previously seropositive from infection or vaccination and immunized with S protein expressing vectors followed by challenge with feline infectious peritonitis virus [73]. However, VAERD has occurred after immunization with SARS inactivated vaccines or alphavirus vectors expressing the nucleocapsid protein. In many instances, inflammatory infiltrates exhibit a Th2 rather than a Th1 phenotype and are characterized by increased numbers of eosinophils [74], [75], [76], [77]. In another study, macaques were immunized with vaccinia virus expressing the SARS protein or passively immunized with plasma from macaques immunized with the same vector. They were then challenged with SARS-CoV. While the animals remained asymptomatic, the nature of the inflammatory infiltrates changed, most prominently from M2- to M1-type macrophages, with increased expression of pro-inflammatory cytokines. Of note, this modification in the immune response did not result in a change in clinical disease [78].
1.2.5. Vaccine-induced enhancement of acquisition of infection
The first large placebo controlled trial (STEP trial) of an Ad5-HIV vaccine candidate was terminated early when a planned interim analysis demonstrated a significantly higher rate of HIV infection in male vaccinees who had been Ad5 seropositive at baseline vs. placebo (5.1% versus 2.2% per year) and/or were uncircumcised (5.2% versus 1.4% per year) [79]. A longer term follow-up analysis (after unblinding) supported the initial finding of enhanced acquisition of HIV infection in the vaccinees compared to the placebo group [80]. Although the difference in the rate of HIV infection was relatively small, it was statistically significant (Hazard ratio of 1.40 (95% CI, 1.03–1.92; p = 0.03)). In an expanded analysis of data, 49 of the 914 male vaccine recipients became HIV infected (annual incidence 4.6%, 95% CI 3.4 to 6.1) and 33 of the 922 male placebo recipients became HIV infected (annual incidence 3.1%, 95% CI 2.1 to 4.3). Potential explanations of this increased susceptibility to HIV infection in the vaccinees included the lack of an HIV-Env antigen in the vaccine, with the possibility that the HIV immune response induced by the vaccine potentially induced attachment of HIV to cellular surface, but without killing or neutralizing the virus, thus enabling viral entry into the cell. While the cause of the apparent vaccine-induced increased susceptibility to acquisition of HIV infection was never fully explained, the conclusion that the vaccine enhanced acquisition of HIV infection remained firm. The clinical data in those vaccinees who were infected were also suggestive of disease enhancement, or at least reduction of the time from acquisition of infection to onset of disease manifestations.
Although no other vaccine has been definitively linked to enhanced acquisition of infection, it is likely that this is not an outcome unique to HIV infection. Enhanced disease associated with inactivated measles or RSV vaccines has become accepted has having occurred, but enhanced acquisition of infection was not widely reported for either the formalin-inactivated RSV and inactivated measles vaccines. However, this may not have been noted due to the high infection rate for measles or RSV in the study populations at the time. Additionally, the four formalin-inactivated RSV vaccine trials were not placebo-controlled [23], [24], [25], [26]. A control vaccine (Parainfluenza 3 vaccine candidate – PIV3) or historical controls were used. Nevertheless, in two of the RSV vaccine trials [23], the attack rates, particularly in infants less than 12 months of age, were higher than in the PIV3 control group. In one study, 23 of 31 (74%) of RSV vaccinated infants later developed RSV infection compared to 21 of 40 (53%) RSV infections in the infants receiving PIV3 (unadjusted chi-square 5.1505; p-value is 0.023) [23]. In a second study, 13 of 43 (30%) RSV vaccinated infants later developed RSV infection compared to 5 of 46 (11%) RSV infections in the infants receiving PIV3 (unadjusted chi-square 5.1645; p-value is 0.023) [24].
In the case of the inactivated measles vaccine, the inactivated measles vaccine was evaluated in trials which compared inactivated vaccine plus at least one dose of live measles vaccine to a live measles vaccine alone control group [81]. Thus, although enhancement of clinical measles was noted in the inactivated measles vaccine group, the number of breakthrough cases of measles was too small to detect a difference in incidence.
Thus, while the occurrence of enhanced acquisition of HIV infection, induced by an Ad5-HIV vaccine candidate became generally acknowledged as a consequence of immunization with that particular vaccine, enhanced acquisition of infection has not been perceived as having occurred with other vaccines. Although the difference in the numbers of HIV infection was relatively small in the STEP trial, the denominators were large and the conclusion that the Ad5-HIV vaccine caused enhancement of acquisition is now generally accepted. For the inactivated RSV vaccine, the trials were relatively small, but the rates of breakthrough RSV infections were much higher and also resulted in statistically significant increases in infection rates in the RSV vaccine groups. This apparent enhanced acquisition of infection may have been overlooked because the severity of enhanced disease occurring in the vaccinees overshadowed the higher risk of RSV infection associated with the vaccine and because the background rates of RSV infection were high in both RSV and PIV3 control vaccines recipients.
1.3. How do existing animal models inform assessment of VAED in humans?
Although animal models have been developed for most respiratory viruses, they rarely reproduce the full spectrum of the corresponding human disease. Therefore, the assessment of the risk of VAED in animal models is imperfect and limited. However, animal models can still provide useful information on potential pathogenic mechanisms and identify markers of potential risks that can be considered for inclusion in clinical trials.
Some lessons may be retained from the previous VAED experience. Today, preclinical RSV vaccine studies that indicate the induction of weak neutralizing and strong non-neutralizing responses with Th2 types of T-cell responses suggesting a risk of VAED may lead to changes in vaccine design strategies. First the selection of antigen(s) is critical to ensure an appropriate balance of neutralizing vs non-neutralizing antibody production. Second, some candidate subunit RSV vaccines are now formulated with Th1-driving adjuvants, which will diminish a prominent Th2 response and an eosinophilic reaction after exposure to the wild virus. Third, preference can be given to RSV vaccines that can induce long-lasting and powerfully neutralizing antibody responses and affinity maturation in order to avoid a gradual waning of antibody levels.
When planning phase 1/2 clinical trials of new candidate vaccines against acute respiratory infections, it is useful to analyze markers of innate and acquired immunity, and observations made in animal models may be informative. Regarding innate immunity, a detailed assessment of monocytes and NK-cell phenotypic markers and various circulating cytokines (e.g. IFN-alpha, IL-10) in the first 24 h post-immunization has been shown to correlate with long term features of subsequent antibody responses [82]. T-cell responses also need to be monitored, including a cytokine profile analysis of both CD4 and CD8 T-cell responses, with multiple Th1 and Th2 markers. The absence of Th2 markers such as IL-4, IL-5 or IL-13, in the presence of consistent IFN-gamma responses may indicate a lower risk of some forms of VAED. Animal models have identified antibody response patterns that are associated with low risk of VAED including a high ratio of neutralizing vs. antigen-binding antibodies anti-receptor binding domain (RBD) antibodies of high affinity (nanomolar range), and antibody kinetics showing sustained IgG responses over time. One may also consider the passive transfer of serum (containing different antibody levels) from immunized Phase 1 trial participants into suitable animal models, prior to viral challenge, to assess the risk of enhanced disease after infection.
1.4. Knowledge gaps in current understanding of potential VAED in the context of SARS-CoV-2
1.4.1. Mechanisms
Various distinct pathways may lead to VAED. SARS-CoV-2 is a novel pathogen facing no specific immunity in populations of all ages and it presents a considerable degree of variability in its clinical manifestations. In addition, its mechanisms of pathogenesis are still unclear. Therefore, understanding how aberrant immune responses may alter a process that is not yet well characterized and that itself presents a considerable range of clinical manifestations is difficult. However, VAED always involves a memory response primed by vaccination and, in the experiences best characterized until now, targets the same organs as wild-type infections. The availability of clinical data and samples from patients with wild-type infections (not vaccinated) is therefore critical to make any assessments and decisions regarding the presence of disease enhancement when analyzing a vaccine candidate.
1.4.2. Animal models
There are a few critical details that should be considered when testing vaccine candidates for the risk of VAED in animal models, regardless of the species selected: [1] the need for a negative wild-type infection control group is paramount. A vaccine may seem safe in the absence of a baseline wild-type infection control, particularly when the VAED positive control presents an exaggerated phenotype; [2] the importance of methodically clearing all control inoculations and challenges from cellular debris, which may enhance reactogenicity in animal models and bias observations; [3] while acknowledging the urgency of the ongoing pandemic, it may be important to wait a considerable period of time between immunization and challenge, since early challenge might prevent investigators from seeing effects that would become obvious later. For this purpose, challenging after antibody titers fall to low – possibly non-protective – levels may be most informative; [4] carefully selecting reproducible models of VAED as positive controls in the evaluation. Some animal models may not exhibit the same manifestations reproducibly and a negative test in the absence of proper positive controls may be deceptive; and [5] prioritize models with clinical manifestations of illness over those exhibiting only pathological changes. Otherwise, results may be distorted by emphasizing pathological differences of uncertain clinical relevance.
1.4.3. Vaccine platforms
Numerous vaccines are under evaluation for SARS-CoV-2 and other emerging pathogens [83]. These include well established vaccine constructs used in existing licensed vaccines (protein subunit, inactivated, virus-like particle, and replicating viral vectored vaccines), and new technologies (nucleic acid, DNA or mRNA-based vaccines) that allow for the rapid development of vaccine candidates [84]. Certain vaccines may be more appropriate for specific populations, such as the elderly, children and pregnant women, compared with healthy adults. The safety, immunogenicity and efficacy of SARS-CoV-2 vaccines must be carefully evaluated prior to their use in the general population, particularly given concerns for disease enhancement and the global need for effective vaccines to control the COVID-19 pandemic.
1.4.4. Adjuvants
Adjuvants have been used in vaccines and given to billions of individuals. Based on this experience and in the context of vaccine development against other pandemic viruses responsible for acute respiratory viral infections, adjuvants may help to: [1] enhance the level and durability of protective humoral response and broaden its epitope-related specificity [2] induce skewed response towards more functional immune responses, including cellular response and [3] generate antigen-sparing approaches able to deliver more vaccine doses in the context of an ongoing pandemic.
Adjuvants have the capacity to increase immune responses through the activation of innate immunity, conditioning the level and quality of antibodies and T-cell responses specific to the vaccine antigen [85]. In addition to antibodies, the most potent adjuvants such as emulsions or those containing saponins and Toll-like receptor ligands have been shown to induce robust and long lasting polyfunctional CD4 + T-cell responses, with a predominant IL-2, IFN-g and TNF response, but remarkably little Th2-associated cytokines [86], [87]. CD8 T-cell responses are also generally not increased by adjuvanted recombinant vaccines.
In the context of disease enhancement, the use of appropriate adjuvants in subunit vaccines may therefore be a possible avenue to manage the potential risk of VAED, in particular those that induce a more potent innate response. However, both pre-existing immunity and the type of antigen influence the impact of the adjuvant on the immune response, and therefore each antigen/adjuvant combination needs to be specifically evaluated. The safety profile of adjuvanted vaccines will also depend on the adjuvant’s mode of action. In early clinical trials of candidate vaccines, it will be important to assess the impact of adjuvant not only on the magnitude, but more critically, on the quality of the immune response, such as antibody functionality and T-cell profiling. In this regard, non-human primates rather than mice have been shown to best reflect the behavior of adjuvants observed in humans and therefore constitutes a good predictive model for formulation selection [88].
1.5. Diagnosis and differential diagnosis of VAED/VAERD
No single or combination of specific confirmatory tests is available to diagnose VAED. As the clinical manifestations of VAED lies within the spectrum of natural disease – occurring more frequently and/or severely in vaccinated individuals – it is also difficult to separate vaccine failure (also called breakthrough disease) from VAED in vaccinated individuals. All cases of vaccine failure should be investigated for VAED. Vaccine failure is defined as the occurrence of the specific vaccine-preventable disease in a person who is appropriately and fully vaccinated, taking into consideration the incubation period and the normal delay for the protection to be acquired as a result of immunization [89]. Assessment of single or multiorgan dysfunction, atypical immune and inflammatory responses, viral identification and quantification, and histopathology may aid in the diagnosis and classification of the extent and severity of disease occurring after vaccination. However, definitive case ascertainment of VAED/VAERD might not be possible, and ascertainment of occurrence of VAED/VAERD might only be feasible in the context of large randomized controlled clinical trials or during post-licensure safety surveillance.
1.6. Disease severity assessment and classification
A classification or a standardized method for the assessment of disease severity are not available for VAED/VAERD. In the case of dengue infection, where the occurrence of antibody-mediated disease enhancement upon reinfection is a well described phenomenon, the existing clinical classification characterizes the more severe manifestations of disease. Similarly, existing clinical disease characterizations and severity of illness scores may be utilized to identify and classify cases of severe or enhanced disease occurring after vaccination.
2. Rationale for selected decisions about the case definition of vaed/vaerd as an adverse event following immunization
In general, VAED is a modified and/or severe presentation of an infectious disease affecting individuals exposed to the wild-type pathogen after having received vaccine designed to prevent infection.
An accepted case definition of VAED does not yet exist. Similarly, harmonized, specific guidance from regulatory bodies regarding the assessment of clinical trial subjects for VAED, is not yet available. A consensus definition is necessary not only in the context of vaccine clinical trials to allow for comparability among different vaccines and clinical studies, but also for the assessment of safety after vaccine licensure and implementation.
2.1. Case definition
2.1.1. Vaccine-associated enhanced disease (VAED)
- 1.Is an illness that occurs in persons who receive a vaccine and who are subsequently infected with the pathogen that the vaccine is meant to protect against. This definition assumes previously antigen-naïve vaccine recipients, which can be assessed by determining seronegative status prior to vaccination, when feasible. The need for documentation of seronegativity prior to vaccination, which can be done retrospectively, is particularly relevant in Phase II-III clinical trials. In the context of such trials, the working group acknowledged the difficulty in distinguishing between vaccine failure and VAED. Thus, all cases of vaccine failure should be evaluated for VAED.
- 2.VAED may present as severe disease or modified/unusual clinical manifestations of a known disease presentation. The illness presumably is more severe or has characteristics that distinguish it from illness that might occur in unvaccinated individuals.
- 3.VAED may involve one or multiple organ systems.
- 4.VAED may also present as an increased incidence of disease in vaccinees compared with controls or known background rates.
2.1.2. Vaccine-associated enhanced respiratory disease (VAERD)
- 5.Refers to the predominant lower respiratory tract presentation of VAED. The mechanisms of pathogenesis might be specific to the lower respiratory tract or part of a systemic process.
2.1.3. Approach for identification of cases of VAED/VAERD
In the context of vaccine clinical trials, the routine collection of adverse events (AE), serious adverse events (SAE), and adverse events of special interest (AESI) is an existing mechanism to evaluate the occurrence of illnesses and outcomes that are serious, including those that are new, require medical care, result in disability, are life threating or result in hospitalization or death. Similarly, AEs are evaluated for severity, using existing tools, such as severity grading scales and toxicity tables for clinical and laboratory outcomes that are adapted to various populations including adults, children and pregnant women [DAIDS Toxicity tables https://rsc.niaid.nih.gov/sites/default/files/daidsgradingcorrectedv21.pdf]. The working group concurs that these methods of assessment of events occurring after vaccination are appropriate to identify triggers that point towards potential cases of VAED/VAERD. Potential cases may be initially identified though clinical characteristics alone (Table 1 ), or complemented with laboratory evaluation (Table 2 ).
Table 1
Factors to consider in the assessment of the clinical presentation of VAED and VAERD.
| A | • Recognizing VAED in an individual patient is particularly challenging.VAED might be identified first as a vaccine failure. The clinical presentation may be variable within a spectrum of disease that ranges from mild to severe, life threatening, with or without long term sequelae, to fatal. |
| B | • Identification of VAED requires the recognition of a clinical presentation that is different, atypical, modified or more severe in comparison to the natural or known (typical) disease presentation, or that occurs at a higher frequency from the control group or expected background rates in the specific target population.• No clinical presentation is pathognomonic for VAED. |
| C | • Identification requires that the clinical syndrome is new or distinct from the typical presentation or from other known diseases, similar or associated disorders, or that such clinical syndrome occurs at higher frequency from the control group or expected background rates. For example, acute respiratory distress syndrome (ARDS) is a distinct entity characterized by rapid and progressive inflammatory changes in the lung parenchyma, resulting in respiratory failure. Diagnosis is based on clinical characteristics and documentation of hypoxemia using accepted standardized definitions (eg. Berlin classification of ARDS). ARDS may occur as a result of a variety of insults that cause inflammation, alveolar cell injury, surfactant dysfunction, and other vascular and hematologic abnormalities, including SARS-CoV-2 infection. ARDS may be a form of clinical presentation of VAED or VAERD. |
| D | • Assessment of the type and frequency of the clinical presentations by developing a clinical profile of cases would be helpful to aid in the more efficient identification of cases through the development of algorithms [90]. |
| E | • Grading of clinical manifestations of disease based on severity using a standardized and/or validated severity of illness score to evaluate all cases is recommended. Several tools are utilized in clinical practice for the assessment of disease severity in adults and children. Commonly used and practical scoring tools for adults are shown in Appendix B, and for children in Appendix C. Appropriate tools for the assessment of severity in various settings (eg. community vs. hospitalized cases) should be selected and used consistently [91].• Whenever feasible, the same clinical scoring tool should be used across related studies and validated. |
Table 2
Assessment for VAED in the context of vaccine development: relevant clinical and laboratory diagnostic parameters.
| Organ system | Clinical parameters | Laboratory parameters |
|---|---|---|
| Respiratory system | ● Cough● Tachypnea● Dyspnea● Lower respiratory tract disease● Respiratory failure● Pulmonary hemorrhage● Radiographic abnormalities | ● Oxygen requirement● Hypoxemia● PaO2● PaO2/FiO2 ratio● Aa gradient |
| Cardiovascular system | ● Tachycardia● Hypotension/ Hypertension● Acute cardiac injury● Vasculitis/ Vasculopathy● Myocarditis● Heart failure● Cardiogenic shock | ● Abnormal ECG● Abnormal Echocardiogram● Troponin● B-Natriuretic Peptide (BNP) |
| Hematopoietic and Immune system | ● Coagulopathy● Disseminated intravascular coagulation● Bleeding/ Thrombotic events | ● Leukopenia, lymphopenia● Thrombocytopenia● B and T cell function assays● Altered coagulation parameters (PT, PTT, D-Dimer, INR) |
| Inflammatory markers | ● Pro-inflammatory state | ● Elevated inflammatory markers (CRP, procalcitonin)● Elevated Ferritin, LDH● Elevated cytokines |
| Renal system | ● Renal dysfunction● Acute kidney injury● Renal replacement therapy | ● Decreased urine output● Serum creatinine● Glomerular filtration rate |
| Gastrointestinal and hepatic system | ● Emesis/Diarrhea● Abdominal pain● Hematochezia/Melena● Hepatitis● Liver dysfunction● Acute liver failure | ● Electrolyte abnormalities● Elevation of liver enzymes● Elevated bilirubin |
| Central Nervous System | ● Altered mental status● Convulsions/seizures● Cranial nerve involvement● Unconsciousness | ● Elevated intracranial pressure● Abnormal CSF parameters |
| Other | ● Fatigue● Myalgia/myositis/myonecrosis● Arthralgia/arthritis● Multiorgan failure● Death | ● Viral load (PCR Ct value)● Antibody titers● Histopathology |
2.1.4. Identification of VAED/VAERD in clinical trials
Identifying cases of VAED/VAERD might be impossible when assessing individual patients, however, in clinical studies, a control group is helpful to compare the frequency of cases and the severity of illness in vaccinees vs. controls, including the occurrence of specific events of concern such as hospitalization and mortality. A comparison group of unvaccinated subjects or active comparator control group is particularly important when background rates of the outcome of interest are not available in the target population. If a control group is not available, comparisons should be made to the expected background rate of the event of interest when it occurs after natural disease in an unvaccinated population. When available, background rates of specific clinical manifestations and outcomes should be used to compare frequencies. Participation of an epidemiologist and statistician is recommended in clinical trial design. Given that the background rate of VAED is unknown, study sample size calculations in early phases of vaccine evaluation should not be based on the occurrence of VAED/VAERD. However, in large studies and in post-implementation phases, reliable surveillance systems should be in place for the timely detection of potential cases, using estimates of defined specific outcomes based on expected background rates or control group rates. All cases of vaccine failure should be evaluated for the possibility of VAED/VAERD, but not all cases of VAED/VAERD will represent vaccine failures. When feasible, a thorough evaluation for alternative etiologies should be conducted, and an adjudication committee, or a safety monitoring committee, or independent expert consultation should be convened to evaluate potential cases.
2.2. Factors to consider in the ascertainment of a case of VAED/VAERD and levels of Diagnostic certainty are described in Table 3
Table 3
Factors to consider in the ascertainment of a case of VAED/VAERD and Levels of Diagnostic Certainty.
| Background rates | Background rates of specific relevant conditions and outcomes, including hospitalization and mortality should be used when available. Backgrounds rates appropriate to the study population and contemporary to the vaccine evaluation should be used. This information might be unavailable or difficult to obtain. Alternatively, assessment of the frequency of events in a control group of unvaccinated individuals is necessary to ascertain the occurrence of events suggestive of VAED or VAERD. Whenever feasible, it is also important to distinguish VAED or VAERD from vaccine failure (as previously defined). |
| Age and gender | The expected severity of outcomes by age group must be described. This is an important factor to consider given that a different clinical presentation from what is expected for a specific age group could be considered VAED or VAERD. When pertinent, gender differences should be considered. |
| Time of onset after vaccination and after infection | VAED or VAERD may occur at any time after vaccination. The timing of occurrence of clinical manifestations of VAED or VAERD after vaccination will be dependent on the mechanism or pathophysiologic pathway leading to disease enhancement after natural infection. VAED or VAERD may present within 2–4 weeks of natural infection, if the expected initial antibody responses are inadequate; or may present at a later time (>1 month or longer) after natural infection if antibody waning is noted or if the mechanism is not exclusively antibody mediated. |
| Duration of follow up | The working group recommends that prolonged follow up is established, at least one year after vaccination or more, depending on the epidemiology of the disease, followed by population-based surveillance in the post-licensure period. In addition to taking into consideration what is realistic in the context of a clinical trial, it is important to consider the circulation of the target pathogen.In the case of endemic, ongoing active circulation, there is a possibility of exposure at any time after vaccination, which requires close follow up immediately after vaccination and potentially, for a prolonged period, depending on the risk of exposure. When pathogens exhibit a seasonal circulation, exposure can be identified through seasonal surveillance and may include follow up for a period of at least one, and preferably two, or more seasons, depending on the pathogen. In cases of sporadic circulation, the exposure periods may be unknown, and the follow up period may be prolonged. |
| Clinical course and progression of symptoms | The following outcomes would be concerning for VAED or VAERD in a person with confirmed infection:a.Death. This would be particularly concerning if death occurs in person without other risk factors for mortality (note phase I-II trials with selected healthy population) or if it occurs at higher rates than expected.b.Hospitalization, including hospitalization above expected rates.c.Worsening or clinical deterioration over time, particularly, although not exclusively, if differing from the anticipated natural course of the disease.d.Prolonged clinical course compared to natural disease.e.Complications of acute disease, new morbidities or new diagnoses subsequent to natural infection post-vaccination (for example higher rate of MIS-C or MIS-A) |
| Control for confounders and comorbidities | In the context of evaluating a case for VAED, it will be important to rule out other infections, comorbidities, drug effects, toxicities, etc. If no alternative explanation for the frequency or severity of illness is identified, including vaccine failure, VAED or VAERD may be considered. |
| Influence of treatment or response to treatment on fulfilment of case definition | Treatment of VAED and VAERD is for the most part, supportive and focused on managing the specific organ dysfunction resulting from the disease. The use of specific antiviral therapy when available, and of immunomodulatory treatments, should be documented. However, the working group considers that treatment or response to treatment is unlikely to be relevant for this case definition as ascertainment is based on comparative clinical severity of illness at presentation. |
| Type of vaccine | Vaccines vary based on the antigen utilized and the addition of adjuvants. At this time, there is insufficient data to determine a priori if any of these platforms is less or more likely to be associated with VAED/VAERD. The working group agrees that it is not possible to know the potential risk for VAED/VAERD of an individual vaccine given various mechanisms leading to disease enhancement, and the different affinity for specific receptors. The use of convalescent sera or monoclonal antibodies might inform potential antibody mediated effect vs. cell mediated mechanisms. |
| Vaccine enhancement vs. vaccine failure | In the event of low/poor vaccine efficacy, infection will occur in vaccinated subjects, with breakthrough disease associated with viral replication. When assessing the safety of a vaccine, there is a need to distinguish between a case where an immune response is not induced from a case where an aberrant non-protective immune response is induced. A thorough assessment of immune responses along with protection from serious disease outcomes is necessary to distinguish enhancement from break-through infection. |
| Geographic and population specific variability in vaccine responses | Other factors including geographic and genetic factors, and individual or population factors such as nutritional status, co-infections, and the effect of co-administration of medications and non-medical products, might also play a role in enhanced disease after vaccination. |
2.3. Diagnostic tests for the assessment of VAED
In addition to clinical parameters and clinical severity of illness grading, the diagnosis of VAED/VAERD should be supported by laboratory, radiographic, and pathology findings, as pertinent. A minimum set of recommended tests to be applied in the assessment of a possible case of VAED based on our current knowledge, is described here and in Table 4 .
Table 4
Suggested laboratory evaluation for the assessment of VAED/VAERD.
| Parameter | Laboratory findings suggestive of VAED/VAERD |
|---|---|
| Evidence inadequate or unbalanced neutralizing antibody responses | • Low or inappropriate total binding (IgG, IgM, IgA) antibody titers• Low neutralizing antibody titers• Low ratio of neutralizing to binding antibody• Low absolute affinity of IgG antibody to receptor binding domain (RBD)• Lack of acquisition or loss of affinity of IgG to RBD• Increased viral load |
| Evidence of inadequate or inappropriately biased cellular immune responses | • Lymphopenia or lymphocytosis• High CD4 lymphocyte subset• Low CD8 lymphocyte subset• Th2 (IL-4, IL-5, IL-13) CD4 T cell predominant response over Th1 (INFg, TNF) responses (testing in vitro stimulation with viral peptides or proteins, ELISPOT, or intracellular cytokine staining assays).• Low virus-specific cytotoxic T-cells (CTL) |
| Evidence of exuberant inflammatory responses | • Elevated IL-1, IL-6, IL-8• Increased pro-inflammatory chemo/cytokines: INF-g, type 1-INF, TNF, CCL2, CCL7• Reduced expression of type I interferons (eg. IFN-α, INF-b)• Elevated C-reactive protein, Ferritin, Lactate dehydrogenase (LDH), D-dimers |
| Evidence of immunopathology in target organs involved, by histopathology | • Present or elevated tissue eosinophils in tissue• Elevated pro-inflammatory Th2 cytokines in tissue (IL4, IL5, IL10, IL13)• C4d tissue deposition (evidence for complement activation through immune complex deposition)• C1q assessments of immune complexes in fluids• Low C3 levels as evidence complement consumption |
2.3.1. Viral identification and quantitation
Confirmation of viral infection by detection and quantitation of virus in specific sites is recommended. These include blood, the upper and lower respiratory tracts, tissue, and other pertinent sterile sites. Characterization of the virus should be performed when feasible (e.g., wild-type vs. vaccine virus, sequencing, emergence of mutations, etc.) Viral quantitation findings should be compared to the extent of observed clinical disease and assessed for consistency.
2.3.2. Immune responses
Evaluation of the immune response after vaccination and at the time of infection could inform the ascertainment of VAED. Whenever feasible, the immune responses should be compared to the expected immune response after natural infection or vaccination. Assessment of neutralizing and total antibody against specific epitopes/targets (for SARS-CoV-2, S and NP) as well as T-cell responses is recommended. Further studies of antibody neutralization, affinity and other stimulation and proliferation assays could be helpful to characterize the immune response.
2.3.3. Antibodies
Several characteristics can assist in exploring the potential risk of VAED in vaccine candidates during early clinical trials. After immunization, the kinetics of the response (sustained vs. peak-valley), neutralization titers, the ratio of neutralizing to S-binding antibodies, and both the absolute affinity for IgG against the RBD compared to that observed in IgG after wild-type infection and the progressive acquisition of affinity for RBD over time, may inform about the quality of antibodies elicited by the immunogens. After infection, the best information to define the potential for VAED when exploring antibody-mediated injury is, when possible, obtaining a biopsy of affected tissues or surrogate materials (e.g., from a small skin biopsy in atypical measles to a nasopharyngeal aspirate in respiratory diseases) that allows detection of C4d deposition as evidence for complement activation through immune complex deposition, C1q assessments of immune complexes in fluids, and C3 levels to explore complement consumption. All these determinations are particularly useful when matched against control samples from subjects experiencing wild-type disease.
2.3.4. Cell mediated immunity
Cell mediated immunity may be assessed by measurement of cell counts to determine the presence of lymphopenia or lymphocytosis, and quantification of specific cell subtypes, such as CD4 and CD8 T-cells. Functional assays will provide information about a change from a Th1 to a Th2 CD4 T-cell response. These assays will measure Th1 (IFN gamma, TNF) vs. Th2 (IL-4, IL-5, IL-13) patterns of response after in vitro stimulation with viral peptides or proteins, in ELISPOT or intracellular cytokine staining assays.
2.3.5. Serum cytokines and other markers
Cytokines are molecules which are secreted by a multitude of cells and effect other cells. They are divided into broad categories: 36 different types of interleukins; 17 types of interferons; 48 chemokines, and 17 members of TNF family at the current time, and more being identified on a regular basis. These cytokines affect growth, maturation, differentiation, regulation and chemotaxis of cells. Cytokines may be biomarkers of VAED and part of the mechanistic process.
Cytokines can also be used as marker of viral disease process and worsening infection. For example, in a severe dengue virus infection, a cytokine storm develops with increased levels of IL-6, IL-8, IFN-α and IFN-γ [92]. Therefore, measuring these cytokines might indicate a worsening of infection. These cytokines or a subset (IL-6 and IL-8) could also be elevated if a VAED-dependent cytokine storm is developing. In ADE seen after viral infections or vaccines, decreased antiviral activity with reduced expression of IFN-α [93] or evidence of worsening virus infection with high titers or increased pro-inflammatory cytokines may be seen. Skewing to Th2 cytokines (IL-4, IL-5, IL-13) and associated eosinophilia may occur as seen in RSV-associated VAED [31]. In the murine model of RSV, TNF-α and IFN-γ are necessary to induce this cytokine storm as other possible biomarkers [94]. Currently, no cytokine/chemokine ‘signature’ associated with VAED has been defined and variability would be expected with different mechanisms of VAED.
2.3.6. Inflammatory responses
A basic assessment of host immune responses after infection should include the evaluation of total white blood cell count and subpopulations (e.g. lymphocyte count, lymphocyte subtypes such as CD8 or CD4), and measurement of inflammatory markers such as C-reactive protein (CRP), Ferritin, Lactate dehydrogenase (LDH), D-dimers, and other specific cytokines (e.g., IL-1, IL-6).
2.3.7. Histopathology (if available)
If available, tissue obtained by biopsy from affected organs or autopsy should be evaluated for evidence of immunopathology.
2.3.8. Radiographic findings
Atypical or more severe involvement of the lower respiratory tract would be anticipated in patients with VAERD. Chest computed tomography (CT) has a high sensitivity for diagnosis of lower respiratory tract disease involvement, including for COVID-19 [95], [96], [97], [98]. A standardized reporting system has been proposed for patients with suspected COVID-19 infection by means of the “CO-RADS classification”, integrating CT findings with clinical symptoms and duration of disease (https://radiologyassistant.nl/chest/covid-19-corads-classification,https://www.rad2share.com/covid-19-ct-report-template).Go to:
3. Case definition of vaccine associated enhanced disease (VAED)
The case definition of VAED is described in Table 5 .
Table 5
Case definition and Levels of Certainty of Vaccine Associated Enhanced Disease.
| LEVEL 1 of Diagnostic Certainty (Definitive case) The working group considers that a Definitive Case (LOC 1) of VAED cannot be ascertained with current knowledge of the mechanisms of pathogenesis of VAED. |
| LEVEL 2 of Diagnostic Certainty (Probable) Rationale for level 2: Ascertainment is based on confirmed infection, with known (2A, higher level of certainty) or without previously known (2B, lower certainty) serostatus, clinical and epidemiologic criteria, and available histopathology. |
| LEVEL 2A. A probable case of VAED is defined by the occurrence of disease in a previously seronegative vaccinated individual with: Laboratory confirmed infection with the pathogen targeted by the vaccine AND Clinical findings of disease involving one or more organ systems (a case of VAERD if the lung is the primarily affected organ) AND Severe disease as evaluated by a clinical severity index/score (systemic in VAED or specific to the lungs in VAERD) AND Increased frequency of severe outcomes (including severe disease, hospitalization and mortality) when compared to a non-vaccinated population (control group or background rates) AND Evidence of immunopathology in target organs involved by histopathology, when available, including any of the following:• Present or elevated tissue eosinophils in tissue• Elevated pro-inflammatory Th2 cytokines in tissue (IL4, IL5, IL10, IL13)• C4d tissue deposition (evidence for complement activation through immune complex deposition)• C1q assessments of immune complexes in fluids• Low C3 levels as evidence complement consumptionAND No identified alternative etiology |
| LEVEL 2B. A probable case of VAED is defined by the occurrence of disease in a vaccinated individual with no prior history of infection and unknown serostatus, with: Laboratory confirmed infection with the pathogen targeted by the vaccine AND Clinical findings of disease involving one or more organ systems (a case of VAERD if the lung is the primarily affected organ) AND Severe disease as evaluated by a clinical severity index/score (systemic in VAED or specific to the lungs in VAERD) AND Increased frequency of severe outcomes (including severe disease, hospitalization and mortality) when compared to a non-vaccinated population (control group or background rates) AND Evidence of immunopathology in target organs involved by histopathology, if available, including any of the following:• Present or elevated tissue eosinophils in tissue• Elevated pro-inflammatory Th2 cytokines in tissue (IL4, IL5, IL10, IL13)• C4d tissue deposition (evidence for complement activation through immune complex deposition)• C1q assessments of immune complexes in fluids• Low C3 levels as evidence complement consumptionAND No identified alternative etiology |
| LEVEL 3 of Diagnostic Certainty (Possible) Rationale for level 3: Ascertainment is based on confirmed or suspected infection, known (3A higher level of certainty) or unknown (3B lower level of certainty) serostatus, clinical and epidemiologic criteria, but no histopathology findings. |
| LEVEL 3A. A possible case of VAED is defined by the occurrence of disease in a previously seronegative vaccinated individual with: Laboratory confirmed infection with the pathogen targeted by the vaccine AND Clinical findings of disease involving one or more organ systems (a case of VAERD if the lung is the primarily affected organ) AND Severe disease as evaluated by a clinical severity index/score (systemic in VAED or specific to the lungs in VAERD) AND Increased frequency of severe outcomes (including severe disease, hospitalization and mortality) when compared to a non-vaccinated population (control group or background rates) AND No identified alternative etiology |
| LEVEL 3B. A possible case of VAED is defined by the occurrence of disease in vaccinated individual with no prior history of infection and unknown serostatus, with: Laboratory confirmed infection with the pathogen targeted by the vaccine AND Clinical findings of disease involving one or more organ systems (a case of VAERD if the lung is the primarily affected organ) AND Severe disease as evaluated by a clinical severity index/score (systemic in VAED or specific to the lungs in VAERD) AND Increased frequency of severe outcomes (including severe disease, hospitalization and mortality) when compared to a non-vaccinated population (control group or background rates) AND No identified alternative etiology |
Open in a separate windowGo to:
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: F. M. Munoz is a consultant for the Coalition for Epidemic Preparedness Innovations (CEPI) for the development of Brighton Collaboration Case Definitions for the Safety Platform for Emergency vACcines (SPEAC) Project.
J. Cramer is a member of the Coalition for Epidemic Preparedness Innovations (CEPI).
C. Dekker is a consultant for the Coalition for Epidemic Preparedness Innovations (CEPI) via Brighton Collaboration for the Safety Platform for Emergency vACcines (SPEAC) Project and a consultant for Medicago Inc.
M. Dudley is a consultant for the Coalition for Epidemic Preparedness Innovations (CEPI) via Brighton Collaboration, for the Safety Platform for Emergency vACcines (SPEAC) Project.
B. Graham is inventor on pending patent applications related to coronavirus vaccines and monoclonal antibodies.
M. Gurwith is a consultant for the Coalition for Epidemic Preparedness Innovations (CEPI) via Brighton Collaboration for the Safety Platform for Emergency vACcines (SPEAC) Project; and is Chief Medical Officer for Verndari, Inc., which is developing vaccines for influenza and Covid-19.
S. Perlman is a member of the ACIP COVID-19 Vaccine Work Group.
F. Polack is an investigator in the Pfizer’s COVID-19 vaccine trial.
B. Ward is Chief Medical Officer for Medicago Inc., which is developing vaccines for influenza and Covid-19.
PH Lambert is a consultant for the Coalition for Epidemic Preparedness Innovations (CEPI) for the development of Case Definitions in the Brighton Collaboration format and for DSMB members training. He is also member of several vaccine DSMBs.
The following authors have no conflict of interests to disclose: J. Spergel, B. Law, E. Van Braeckel, A. Didierlaurent.Go to:
Acknowledgements
The authors are grateful for the support and helpful comments provided by the Brighton Collaboration Reference group who provided peer review, the expert reviewers of the final draft of the manuscript, Kathy Edwards, Kanta Subbarao and Neil Halsey, as well as members of the Safety Platform for Emergency Vaccines and other experts consulted as part of the process, including Stephen Evans, Steve Black, Svein R. Andersen, Miriam Sturkenboom, Wan-Ting Huang, and Robert Chen.
We acknowledge the financial support provided by the Coalition for Epidemic Preparedness Innovations (CEPI) for our work under a service order entitled Safety Platform for Emergency vACcines (SPEAC) Project with the Brighton Collaboration, a program of the Task Force for Global Health, Decatur, GA.
Disclaimer
The findings, opinions and assertions contained in this consensus document are those of the individual scientific professional members of the working group. They do not necessarily represent the official positions of each participant’s organization (e.g., government, university, or corporation). Specifically, the findings and conclusions in this paper are those of the authors and do not necessarily represent the views of their respective institutions.Go to:
Footnotes
Appendix ASupplementary data to this article can be found online at https://doi.org/10.1016/j.vaccine.2021.01.055.Go to:
Appendix A. Supplementary material
The following are the Supplementary data to this article:Supplementary data 1:Click here to view.(22K, docx)
Supplementary data 2:Click here to view.(277K, docx)
Supplementary data 3:Click here to view.(615K, docx)
Supplementary data 4:Click here to view.(31K, docx)Go to:
References
1. Graham B.S. Rapid COVID-19 vaccine development. Science (New York, NY) 2020;368(6494):945–946. [PubMed] [Google Scholar]2. Acosta P.L., Caballero M.T., Polack F.P. Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease. Clin Vaccine Immunol: CVI. 2015;23(3):189–195. [PMC free article] [PubMed] [Google Scholar]3. Jorquera P.A., Anderson L., Tripp R.A. Understanding respiratory syncytial virus (RSV) vaccine development and aspects of disease pathogenesis. Expert Rev Vaccines. 2016;15(2):173–187. [PubMed] [Google Scholar]4. Vittucci A.C., Zangari P., Ciarlitto C., Di Camillo C., Grandin A., Cotugno N. Active prophylaxis for respiratory syncytial virus: current knowledge and future perspectives. Minerva Pediatr. 2018;70(6):566–578. [PubMed] [Google Scholar]5. Moss W.J., Polack F.P. Immune responses to measles and measles vaccine: challenges for measles control. Viral Immunol. 2001;14(4):297–309. [PubMed] [Google Scholar]6. Griffin D.E., Pan C.H., Moss W.J. Measles vaccines. Front Biosci: J Virtual Library. 2008;13:1352–1370. [PubMed] [Google Scholar]7. Wilder-Smith A., Ooi E.E., Horstick O., Wills B. Dengue. Lancet (London, England) 2019;393(10169):350–363. [PubMed] [Google Scholar]8. Gan E.S., Ting D.H., Chan K.R. The mechanistic role of antibodies to dengue virus in protection and disease pathogenesis. Expert Rev Anti-Infective Therapy. 2017;15(2):111–119. [PubMed] [Google Scholar]9. Halstead S.B., Russell P.K. Protective and immunological behavior of chimeric yellow fever dengue vaccine. Vaccine. 2016;34(14):1643–1647. [PubMed] [Google Scholar]10. Srikiatkhachorn A., Yoon I.K. Immune correlates for dengue vaccine development. Expert Rev Vaccines. 2016;15(4):455–465. [PMC free article] [PubMed] [Google Scholar]11. Wan S.W., Lin C.F., Yeh T.M., Liu C.C., Liu H.S., Wang S. Autoimmunity in dengue pathogenesis. J Formosan Med Assoc = Taiwan yi zhi. 2013;112(1):3–11. [PubMed] [Google Scholar]12. Rajao D.S., Anderson T.K., Gauger P.C., Vincent A.L. Pathogenesis and Vaccination of Influenza A Virus in Swine. In: Compans R.W., Oldstone M.B.A., editors. Influenza Pathogenesis and Control – Volume 1. Current Topics in Microbiology and Immunology. 1 ed. Springer International Publishing; Switzerland: 2014. pp. 315–320. [Google Scholar]13. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J. A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England J Med. 2020;382(8):727–733. [PMC free article] [PubMed] [Google Scholar]14. Agrawal A.S., Tao X., Algaissi A., Garron T., Narayanan K., Peng B.H. Immunization with inactivated Middle East Respiratory Syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Human Vaccines Immunotherap. 2016;12(9):2351–2356. [PMC free article] [PubMed] [Google Scholar]15. Wu Z., McGoogan J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020 [PubMed] [Google Scholar]16. Martines R.B., Ritter J.M., Matkovic E., Gary J., Bollweg B.C., Bullock H. Pathology and Pathogenesis of SARS-CoV-2 Associated with Fatal Coronavirus Disease, United States. Emerg Infect Dis. 2020;26(9) [PMC free article] [PubMed] [Google Scholar]17. Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respiratory Med. 2020;8(4):420–422. [PMC free article] [PubMed] [Google Scholar]18. The Royal College of Paediatrics and Child Health. Guidance – Paediatric multisystem inflammatory syndrome temporally associated with COVID-19 (PIMS); 2020 [Available from: https://www.rcpch.ac.uk/resources/guidance-paediatric-multisystem-inflammatory-syndrome-temporally-associated-covid-19-pims].19. Riphagen S., Gomez X., Gonzalez-Martinez C., Wilkinson N., Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet (London, England) 2020;395(10237):1607–1608. [PMC free article] [PubMed] [Google Scholar]20. Verdoni L., Mazza A., Gervasoni A., Martelli L., Ruggeri M., Ciuffreda M. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet (London, England) 2020;395(10239):1771–1778. [PMC free article] [PubMed] [Google Scholar]21. Centers for Disease Control and Prevention. Multisystem Inflammatory Syndrome in Children (MIS-C) Associated with Coronavirus Disease 2019 (COVID-19); 2020 [updated March 27, 2020. Available from: https://emergency.cdc.gov/han/2020/han00432.asp.22. Lambert P.-H., Ambrosino D.M., Andersen S.R., Baric R.S., Black S.B., Chen R.T. Consensus summary report for CEPI/BC March 12–13, 2020 meeting: Assessment of risk of disease enhancement with COVID-19 vaccines. Vaccine. 2020;38(31):4783–4791. [PMC free article] [PubMed] [Google Scholar]23. Kim H.W., Canchola J.G., Brandt C.D., Pyles G., Chanock R.M., Jensen K. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89(4):422–434. [PubMed] [Google Scholar]24. Chin J., Magoffin R.L., Shearer L.A., Schieble J.H., Lennette E.H. Field evaluation of a respiratory syncytial virus vaccine and a trivalent parainfluenza virus vaccine in a pediatric population. Am J Epidemiol. 1969;89(4):449–463. [PubMed] [Google Scholar]25. Fulginiti V.A., Eller J.J., Sieber O.F., Joyner J.W., Minamitani M., Meiklejohn G. Respiratory virus immunization. I. A field trial of two inactivated respiratory virus vaccines; an aqueous trivalent parainfluenza virus vaccine and an alum-precipitated respiratory syncytial virus vaccine. Am J Epidemiol. 1969;89(4):435–448. [PubMed] [Google Scholar]26. Kapikian A.Z., Mitchell R.H., Chanock R.M., Shvedoff R.A., Stewart C.E. An epidemiologic study of altered clinical reactivity to respiratory syncytial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am J Epidemiol. 1969;89(4):405–421. [PubMed] [Google Scholar]27. Polack F.P., Teng M.N., Collins P.L., Prince G.A., Exner M., Regele H. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med. 2002;196(6):859–865. [PMC free article] [PubMed] [Google Scholar]28. Delgado M.F., Coviello S., Monsalvo A.C., Melendi G.A., Hernandez J.Z., Batalle J.P. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. 2009;15(1):34–41. [PMC free article] [PubMed] [Google Scholar]29. Connors M., Kulkarni A.B., Firestone C.Y., Holmes K.L., Morse H.C., 3rd, Sotnikov A.V. Pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of formalin-inactivated RSV-immunized BALB/c mice is abrogated by depletion of CD4+ T cells. J Virol. 1992;66(12):7444–7451. [PMC free article] [PubMed] [Google Scholar]30. Killikelly A.M., Kanekiyo M., Graham B.S. Pre-fusion F is absent on the surface of formalin-inactivated respiratory syncytial virus. Sci Rep. 2016;6:34108. [PMC free article] [PubMed] [Google Scholar]31. Boukhvalova M.S., Prince G.A., Soroush L., Harrigan D.C., Vogel S.N., Blanco J.C. The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine. 2006;24(23):5027–5035. [PubMed] [Google Scholar]32. Knudson C.J., Hartwig S.M., Meyerholz D.K., Varga S.M. RSV vaccine-enhanced disease is orchestrated by the combined actions of distinct CD4 T cell subsets. PLoS Pathog. 2015;11(3) [PMC free article] [PubMed] [Google Scholar]33. Castilow EM, Meyerholz DK, Varga SM. IL-13 is required for eosinophil entry into the lung during respiratory syncytial virus vaccine-enhanced disease. J Immunol (Baltimore, Md: 1950) 2008;180(4):2376–84. [PubMed]34. Openshaw P.J., Tregoning J.S. Immune responses and disease enhancement during respiratory syncytial virus infection. Clin Microbiol Rev. 2005;18(3):541–555. [PMC free article] [PubMed] [Google Scholar]35. Graham BS, Henderson GS, Tang YW, Lu X, Neuzil KM, Colley DG. Priming immunization determines T helper cytokine mRNA expression patterns in lungs of mice challenged with respiratory syncytial virus. J Immunol (Baltimore, Md: 1950) 1993;151(4):2032–40. [PubMed]36. Connors M., Giese N.A., Kulkarni A.B., Firestone C.Y., Morse H.C., 3rd, Murphy B.R. Enhanced pulmonary histopathology induced by respiratory syncytial virus (RSV) challenge of formalin-inactivated RSV-immunized BALB/c mice is abrogated by depletion of interleukin-4 (IL-4) and IL-10. J Virol. 1994;68(8):5321–5325. [PMC free article] [PubMed] [Google Scholar]37. Stokes K.L., Currier M.G., Sakamoto K., Lee S., Collins P.L., Plemper R.K. The respiratory syncytial virus fusion protein and neutrophils mediate the airway mucin response to pathogenic respiratory syncytial virus infection. J Virol. 2013;87(18):10070–10082. [PMC free article] [PubMed] [Google Scholar]38. Dakhama A, Park JW, Taube C, Joetham A, Balhorn A, Miyahara N, et al. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J Immunol (Baltimore, Md: 1950) 2005;175(3):1876-83. [PubMed]39. Katz S.L., John F. Enders and measles virus vaccine–a reminiscence. Curr Top Microbiol Immunol. 2009;329:3–11. [PubMed] [Google Scholar]40. Katz S.L., Kempe C.H., Black F.L., Lepow M.L., Krugman S., Haggerty R.J. Studies on an attenuated measles-virus vaccine. VIII. General summary and evaluation of the results of vaccine. New England J Med. 1960;263:180–184. [PubMed] [Google Scholar]41. Rød T., Haug K.W., Ulstrup J.C. Atypical measles after vaccination with killed vaccine. Scand J Infect Dis. 1970;2(3):161–165. [PubMed] [Google Scholar]42. Young LW, Smith DI, Glasgow LA. Pneumonia of atypical measles. Residual nodular lesions. Am J Roentgenol, Radium Therapy, Nucl Med 1970;110(3):439-48. [PubMed]43. Polack F.P., Auwaerter P.G., Lee S.H., Nousari H.C., Valsamakis A., Leiferman K.M. Production of atypical measles in rhesus macaques: evidence for disease mediated by immune complex formation and eosinophils in the presence of fusion-inhibiting antibody. Nat Med. 1999;5(6):629–634. [PubMed] [Google Scholar]44. Polack F.P., Hoffman S.J., Crujeiras G., Griffin D.E. A role for nonprotective complement-fixing antibodies with low avidity for measles virus in atypical measles. Nat Med. 2003;9(9):1209–1213. [PubMed] [Google Scholar]45. Polack F.P., Lee S.H., Permar S., Manyara E., Nousari H.G., Jeng Y. Successful DNA immunization against measles: neutralizing antibody against either the hemagglutinin or fusion glycoprotein protects rhesus macaques without evidence of atypical measles. Nat Med. 2000;6(7):776–781. [PubMed] [Google Scholar]46. Erlenhoefer C., Wurzer W.J., Löffler S., Schneider-Schaulies S., ter Meulen V., Schneider-Schaulies J. CD150 (SLAM) is a receptor for measles virus but is not involved in viral contact-mediated proliferation inhibition. J Virol. 2001;75(10):4499–4505. [PMC free article] [PubMed] [Google Scholar]47. Krause P.J., Cherry J.D., Naiditch M.J., Deseda-Tous J., Walbergh E.J. Revaccination of previous recipients of killed measles vaccine: clinical and immunologic studies. J Pediatrics. 1978;93(4):565–571. [PubMed] [Google Scholar]48. Moghaddam A., Olszewska W., Wang B., Tregoning J.S., Helson R., Sattentau Q.J. A potential molecular mechanism for hypersensitivity caused by formalin-inactivated vaccines. Nat Med. 2006;12(8):905–907. [PubMed] [Google Scholar]49. Loebbermann J., Durant L., Thornton H., Johansson C., Openshaw P.J. Defective immunoregulation in RSV vaccine-augmented viral lung disease restored by selective chemoattraction of regulatory T cells. Proc Natl Acad Sci USA. 2013;110(8):2987–2992. [PMC free article] [PubMed] [Google Scholar]50. Srikiatkhachorn A., Braciale T.J. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J Exp Med. 1997;186(3):421–432. [PMC free article] [PubMed] [Google Scholar]51. Hussell T., Baldwin C.J., O’Garra A., Openshaw P.J. CD8+ T cells control Th2-driven pathology during pulmonary respiratory syncytial virus infection. Eur J Immunol. 1997;27(12):3341–3349. [PubMed] [Google Scholar]52. Castilow EM, Legge KL, Varga SM. Cutting edge: Eosinophils do not contribute to respiratory syncytial virus vaccine-enhanced disease. J Immunol (Baltimore, Md: 1950) 2008;181(10):6692-6. [PMC free article] [PubMed]53. Halstead S.B. Pathogenesis of dengue: challenges to molecular biology. Science (New York, NY) 1988;239(4839):476–481. [PubMed] [Google Scholar]54. Halstead S.B., O’Rourke E.J. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med. 1977;146(1):201–217. [PMC free article] [PubMed] [Google Scholar]55. Katzelnick L.C., Gresh L., Halloran M.E., Mercado J.C., Kuan G., Gordon A. Antibody-dependent enhancement of severe dengue disease in humans. Science (New York, NY) 2017;358(6365):929–932. [PMC free article] [PubMed] [Google Scholar]56. Hadinegoro S.R., Arredondo-García J.L., Capeding M.R., Deseda C., Chotpitayasunondh T., Dietze R. Efficacy and Long-Term Safety of a Dengue Vaccine in Regions of Endemic Disease. New England J Med. 2015;373(13):1195–1206. [PubMed] [Google Scholar]57. Sridhar S., Luedtke A., Langevin E., Zhu M., Bonaparte M., Machabert T. Effect of Dengue Serostatus on Dengue Vaccine Safety and Efficacy. New England J Med. 2018;379(4):327–340. [PubMed] [Google Scholar]58. Britto C., Dold C., Reyes-Sandoval A., Rollier C.S. Rapid travel to a Zika vaccine: are we heading towards success or more questions? Expert Opin Biol Ther. 2018;18(11):1171–1179. [PubMed] [Google Scholar]59. Priyamvada L., Suthar M.S., Ahmed R., Wrammert J. Humoral Immune Responses Against Zika Virus Infection and the Importance of Preexisting Flavivirus Immunity. J Infect Dis. 2017;216(suppl_10):S906–S911. [PMC free article] [PubMed] [Google Scholar]60. Heinz F.X., Stiasny K. The Antigenic Structure of Zika Virus and Its Relation to Other Flaviviruses: Implications for Infection and Immunoprophylaxis. Microbiology and molecular biology reviews. MMBR. 2017;81(1) [PMC free article] [PubMed] [Google Scholar]61. Huang K.J., Su I.J., Theron M., Wu Y.C., Lai S.K., Liu C.C. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol. 2005;75(2):185–194. [PMC free article] [PubMed] [Google Scholar]62. Theron M., Huang K.J., Chen Y.W., Liu C.C., Lei H.Y. A probable role for IFN-gamma in the development of a lung immunopathology in SARS. Cytokine. 2005;32(1):30–38. [PMC free article] [PubMed] [Google Scholar]63. Cameron M.J., Ran L., Xu L., Danesh A., Bermejo-Martin J.F., Cameron C.M. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J Virol. 2007;81(16):8692–8706. [PMC free article] [PubMed] [Google Scholar]64. Wong C.K., Lam C.W., Wu A.K., Ip W.K., Lee N.L., Chan I.H. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. 2004;136(1):95–103. [PMC free article] [PubMed] [Google Scholar]65. Jiang Y., Xu J., Zhou C., Wu Z., Zhong S., Liu J. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med. 2005;171(8):850–857. [PubMed] [Google Scholar]66. Thompson B.T., Chambers R.C., Liu K.D. Acute Respiratory Distress Syndrome. New England J Med. 2017;377(6):562–572. [PubMed] [Google Scholar]67. Cheung C.Y., Poon L.L., Ng I.H., Luk W., Sia S.F., Wu M.H. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79(12):7819–7826. [PMC free article] [PubMed] [Google Scholar]68. Law H.K., Cheung C.Y., Ng H.Y., Sia S.F., Chan Y.O., Luk W. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106(7):2366–2374. [PMC free article] [PubMed] [Google Scholar]69. Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19(2):181–193. [PMC free article] [PubMed] [Google Scholar]70. Lau S.K.P., Lau C.C.Y., Chan K.H., Li C.P.Y., Chen H., Jin D.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J General Virol. 2013;94(Pt 12):2679–2690. [PubMed] [Google Scholar]71. Zielecki F., Weber M., Eickmann M., Spiegelberg L., Zaki A.M., Matrosovich M. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J Virol. 2013;87(9):5300–5304. [PMC free article] [PubMed] [Google Scholar]72. Zhou J., Chu H., Li C., Wong B.H., Cheng Z.S., Poon V.K. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J Infect Dis. 2014;209(9):1331–1342. [PMC free article] [PubMed] [Google Scholar]73. Mahmudpour M., Roozbeh J., Keshavarz M., Farrokhi S., Nabipour I. COVID-19 cytokine storm: The anger of inflammation. Cytokine. 2020;133 [PMC free article] [PubMed] [Google Scholar]74. Bolles M., Deming D., Long K., Agnihothram S., Whitmore A., Ferris M. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol. 2011;85(23):12201–12215. [PMC free article] [PubMed] [Google Scholar]75. Deming D., Sheahan T., Heise M., Yount B., Davis N., Sims A. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 2006;3(12) [PMC free article] [PubMed] [Google Scholar]76. Sheahan T., Whitmore A., Long K., Ferris M., Rockx B., Funkhouser W. Successful vaccination strategies that protect aged mice from lethal challenge from influenza virus and heterologous severe acute respiratory syndrome coronavirus. J Virol. 2011;85(1):217–230. [PMC free article] [PubMed] [Google Scholar]77. Yasui F, Kai C, Kitabatake M, Inoue S, Yoneda M, Yokochi S, et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J Immunol (Baltimore, Md: 1950) 2008;181(9):6337-48. [PubMed]78. Liu L., Wei Q., Lin Q., Fang J., Wang H., Kwok H. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019;4(4) [PMC free article] [PubMed] [Google Scholar]79. Buchbinder S.P., Mehrotra D.V., Duerr A., Fitzgerald D.W., Mogg R., Li D. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet (London, England) 2008;372(9653):1881–1893. [PMC free article] [PubMed] [Google Scholar]80. Duerr A., Huang Y., Buchbinder S., Coombs R.W., Sanchez J., del Rio C. Extended Follow-up Confirms Early Vaccine-Enhanced Risk of HIV Acquisition and Demonstrates Waning Effect Over Time Among Participants in a Randomized Trial of Recombinant Adenovirus HIV Vaccine (Step Study) J Infect Dis. 2012;206(2):258–266. [PMC free article] [PubMed] [Google Scholar]81. Vaccination against measles: clinical trial of live measles vaccine given alone and live vaccine preceded by killed vaccine. Second report to the medical research council by the measles vaccines committee. Brit Med J 1968;2(5603):449-52. [PMC free article] [PubMed]82. Rechtien A., Richert L., Lorenzo H., Martrus G., Hejblum B., Dahlke C. Systems Vaccinology Identifies an Early Innate Immune Signature as a Correlate of Antibody Responses to the Ebola Vaccine rVSV-ZEBOV. Cell Reports. 2017;20(9):2251–2261. [PMC free article] [PubMed] [Google Scholar]83. Thanh Le T., Andreadakis Z., Kumar A., Gómez Román R., Tollefsen S., Saville M. The COVID-19 vaccine development landscape. Nat Rev Drug Discovery. 2020;19(5):305–306. [PubMed] [Google Scholar]84. Baay M., Lina B., Fontanet A., Marchant A., Saville M., Sabot P. SARS-CoV-2: Virology, epidemiology, immunology and vaccine development. Biol: J Int Assoc Biol Stand. 2020;66:35–40. [PMC free article] [PubMed] [Google Scholar]85. O’Hagan D.T., Friedland L.R., Hanon E., Didierlaurent A.M. Towards an evidence based approach for the development of adjuvanted vaccines. Curr Opin Immunol. 2017;47:93–102. [PubMed] [Google Scholar]86. Garçon N., Di Pasquale A. From discovery to licensure, the Adjuvant System story. Human Vaccines Immunotherap. 2017;13(1):19–33. [PMC free article] [PubMed] [Google Scholar]87. Del Giudice G., Rappuoli R., Didierlaurent A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin Immunol. 2018;39:14–21. [PubMed] [Google Scholar]88. Thompson E.A., Loré K. Non-human primates as a model for understanding the mechanism of action of toll-like receptor-based vaccine adjuvants. Curr Opin Immunol. 2017;47:1–7. [PubMed] [Google Scholar]89. Heininger U., Bachtiar N.S., Bahri P., Dana A., Dodoo A., Gidudu J. The concept of vaccination failure. Vaccine. 2012;30(7):1265–1268. [PubMed] [Google Scholar]90. Liang W., Liang H., Ou L., Chen B., Chen A., Li C. Development and Validation of a Clinical Risk Score to Predict the Occurrence of Critical Illness in Hospitalized Patients With COVID-19. JAMA Intern Med. 2020 [PMC free article] [PubMed] [Google Scholar]91. Force* TADT. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA 2012;307(23):2526-33. [PubMed]92. John D.V., Lin Y.S., Perng G.C. Biomarkers of severe dengue disease – a review. J Biomed Sci. 2015;22:83. [PMC free article] [PubMed] [Google Scholar]93. Flipse J., Wilschut J., Smit J.M. Molecular mechanisms involved in antibody-dependent enhancement of dengue virus infection in humans. Traffic (Copenhagen, Denmark) 2013;14(1):25–35. [PubMed] [Google Scholar]94. Walsh K.B., Teijaro J.R., Brock L.G., Fremgen D.M., Collins P.L., Rosen H. Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by sphingosine-1-phosphate 1 receptor agonist therapy. J Virol. 2014;88(11):6281–6293. [PMC free article] [PubMed] [Google Scholar]95. Ai T., Yang Z., Hou H., Zhan C., Chen C., Lv W. Correlation of Chest CT and RT-PCR Testing for Coronavirus Disease 2019 (COVID-19) in China: A Report of 1014 Cases. Radiology. 2020;296(2):E32–E40. [PMC free article] [PubMed] [Google Scholar]96. Shi H., Han X., Jiang N., Cao Y., Alwalid O., Gu J. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. 2020;20(4):425–434. [PMC free article] [PubMed] [Google Scholar]97. Zhang R., Ouyang H., Fu L., Wang S., Han J., Huang K. CT features of SARS-CoV-2 pneumonia according to clinical presentation: a retrospective analysis of 120 consecutive patients from Wuhan city. Eur Radiol. 2020;30(8):4417–4426. [PMC free article] [PubMed] [Google Scholar]98. Rubin G.D., Ryerson C.J., Haramati L.B., Sverzellati N., Kanne J.P., Raoof S. The Role of Chest Imaging in Patient Management during the COVID-19 Pandemic: A Multinational Consensus Statement from the Fleischner Society. Radiology. 2020;296(1):172–180. [PMC free article] [PubMed] [Google Scholar]99. Jones A.E., Trzeciak S., Kline J.A. The Sequential Organ Failure Assessment score for predicting outcome in patients with severe sepsis and evidence of hypoperfusion at the time of emergency department presentation. Crit Care Med. 2009;37(5):1649–1654. [PMC free article] [PubMed] [Google Scholar]100. Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet (London, England) 2020;395(10229):1054–1062. [PMC free article] [PubMed] [Google Scholar]101. Ferreira F.L., Bota D.P., Bross A., Mélot C., Vincent J.-L. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA. 2001;286(14):1754–1758. [PubMed] [Google Scholar]102. Seymour C.W., Liu V.X., Iwashyna T.J., Brunkhorst F.M., Rea T.D., Scherag A. Assessment of clinical criteria for sepsis: for the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315(8):762–774. [PMC free article] [PubMed] [Google Scholar]103. Zhang J., Wang X., Jia X., Li J., Hu K., Chen G. Risk factors for disease severity, unimprovement, and mortality in COVID-19 patients in Wuhan, China. Clin Microbiol Infect: Off Publ Europ Soc Clin Microbiol Infect Dis. 2020;26(6):767–772. [PMC free article] [PubMed] [Google Scholar]104. Monaghan A. Detecting and managing deterioration in children. Paediatric Nursing. 2005;17(1):32–35. [PubMed] [Google Scholar]105. Chapman S.M., Wray J., Oulton K., Peters M.J. Systematic review of paediatric track and trigger systems for hospitalised children. Resuscitation. 2016;109:87–109. [PubMed] [Google Scholar]106. Chapman S.M., Wray J., Oulton K., Pagel C., Ray S., Peters M.J. ‘The Score Matters’: wide variations in predictive performance of 18 paediatric track and trigger systems. Arch Dis Child. 2017;102(6):487–495. [PubMed] [Google Scholar]107. Parshuram C.S., Duncan H.P., Joffe A.R., Farrell C.A., Lacroix J.R., Middaugh K.L. Multicentre validation of the bedside paediatric early warning system score: a severity of illness score to detect evolving critical illness in hospitalised children. Critical Care (London, England) 2011;15(4):R184. [PMC free article] [PubMed] [Google Scholar]108. Parshuram C.S., Dryden-Palmer K., Farrell C., Gottesman R., Gray M., Hutchison J.S. Evaluating processes of care and outcomes of children in hospital (EPOCH): study protocol for a randomized controlled trial. Trials. 2015;16:245. [PMC free article] [PubMed] [Google Scholar]109. McElroy T., Swartz E.N., Hassani K., Waibel S., Tuff Y., Marshall C. Implementation study of a 5-component pediatric early warning system (PEWS) in an emergency department in British Columbia, Canada, to inform provincial scale up. BMC Emergency Med. 2019;19(1):74. [PMC free article] [PubMed] [Google Scholar]110. Agulnik A., Gossett J., Carrillo A.K., Kang G., Morrison R.R. Abnormal Vital Signs Predict Critical Deterioration in Hospitalized Pediatric Hematology-Oncology and Post-hematopoietic Cell Transplant Patients. Front Oncol. 2020;10:354. [PMC free article] [PubMed] [Google Scholar]111. Lu Y., Liu Z., Li S., Wang Y., Li C., Yuan E. [Nested case-control study on paediatric early warning score and ventilator-associated complications in children with acute respiratory distress syndrome]. Zhong nan da xue xue bao Yi xue ban = J Central South Univ Med Sci. 2019;44(9):996–1002. [PubMed] [Google Scholar]112. Pollack M.M., Ruttimann U.E., Getson P.R. Pediatric risk of mortality (PRISM) score. Crit Care Med. 1988;16(11):1110–1116. [PubMed] [Google Scholar]113. El-Mashad G.M., El-Mekkawy M.S., Zayan M.H. Paediatric sequential organ failure assessment (pSOFA) score: a new mortality prediction score in the paediatric intensive care unit. Anales de Pediatría (English Edition) 2020;92(5):277–285. [PubMed] [Google Scholar]114. Typpo K.V., Petersen N.J., Hallman D.M., Markovitz B.P., Mariscalco M.M. Day 1 multiple organ dysfunction syndrome is associated with poor functional outcome and mortality in the pediatric intensive care unit. Pediatr Crit Care Med. 2009;10(5):562–570. [PMC free article] [PubMed] [Google Scholar]115. Wilkinson J.D., Pollack M.M., Ruttimann U.E., Glass N.L., Yeh T.S. Outcome of pediatric patients with multiple organ system failure. Crit Care Med. 1986;14(4):271–274. [PubMed] [Google Scholar]116. Leteurtre S., Martinot A., Duhamel A., Gauvin F., Grandbastien B., Thi Vu.N. Development of a Pediatric Multiple Organ Dysfunction Score: Use of Two Strategies. Med Decis Making. 1999;19(4):399–410. [PubMed] [Google Scholar]117. Leteurtre S., Duhamel A., Salleron J., Grandbastien B., Lacroix J., Leclerc F. PELOD-2: an update of the PEdiatric logistic organ dysfunction score. Crit Care Med. 2013;41(7):1761–1773. [PubMed] [Google Scholar]118. Schlapbach L.J., Straney L., Bellomo R., MacLaren G., Pilcher D. Prognostic accuracy of age-adapted SOFA, SIRS, PELOD-2, and qSOFA for in-hospital mortality among children with suspected infection admitted to the intensive care unit. Intensive Care Med. 2018;44(2):179–188. [PMC free article] [PubMed] [Google Scholar]119. Graciano A.L., Balko J.A., Rahn D.S., Ahmad N., Giroir B.P. The Pediatric Multiple Organ Dysfunction Score (P-MODS): development and validation of an objective scale to measure the severity of multiple organ dysfunction in critically ill children. Crit Care Med. 2005;33(7):1484–1491. [PubMed] [Google Scholar]120. Vincent J., Moreno R., Takala J., Willatts S., De Mendonça A., Bruining H. The SOFA score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996;22(7):707–710. [PubMed] [Google Scholar]121. Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M. The third international consensus definitions for sepsis and septic shock (Sepsis-3) JAMA. 2016;315(8):801–810. [PMC free article] [PubMed] [Google Scholar]122. Matics T.J., Sanchez-Pinto L.N. Adaptation and validation of a pediatric sequential organ failure assessment score and evaluation of the sepsis-3 definitions in critically ill children. JAMA Pediatrics. 2017;171(10):e172352. [PMC free article] [PubMed] [Google Scholar]123. Slater A., Shann F., Pearson G., Group P.S. PIM2: a revised version of the Paediatric Index of Mortality. Intensive Care Med. 2003;29(2):278–285. [PubMed] [Google Scholar]124. Dewi W., Christie C.D., Wardhana A., Fadhilah R., Pardede S.O. Pediatric Logistic Organ Dysfunction-2 (Pelod-2) score as a model for predicting mortality in pediatric burn injury. Ann Burns Fire Disasters. 2019;32(2):135–142. [PMC free article] [PubMed] [Google Scholar]125. Watson RS, Crow SS, Hartman ME, Lacroix J, Odetola FO. Epidemiology and Outcomes of Pediatric Multiple Organ Dysfunction Syndrome. Pediatric Crit Care Med: J Soc Crit Care Med World Federat Pediatric Intensive Crit Care Soc 2017;18(3_suppl Suppl 1):S4-S16. [PMC free article] [PubMed]126. Beltramo F., Khemani R.G. Definition and global epidemiology of pediatric acute respiratory distress syndrome. Ann Trans Med. 2019;7(19):502. [PMC free article] [PubMed] [Google Scholar]127. Singh J. International conference on harmonization of technical requirements for registration of pharmaceuticals for human use. J Pharmacol Pharmacotherap. 2015;6(3):185–187. [Google Scholar]128. The Council for International Organizations of Medical Sciences. CIOMS I FORM; 2020 [Available from: https://cioms.ch/cioms-i-form/].