
- Georgios Divolis, Evgenia Synolaki, Rodoula Tringidou, et.al., Respiratory Research volume 26, Article number: 213 (2025) Cite this article
Abstract
Background
Coronavirus disease 2019 (COVID-19) is a respiratory disease linked with deregulated immune responses, leading to hyperinflammation, acute respiratory distress syndrome, and pulmonary fibrosis, often with fatal outcomes. Neutrophils play a central role in COVID-19 pathogenesis, with elevated peripheral blood neutrophil counts correlating with disease severity. Despite extensive research, the molecular processes associated with neutrophil hyperactivation in COVID-19 remain elusive.
Methods
To investigate the molecular signatures underlying neutrophil-driven pathology, we conducted transcriptome analysis in neutrophils isolated from the peripheral blood of COVID-19 patients versus healthy individuals. To evaluate the specificity of identified neutrophil signatures in COVID-19, we extended our transcriptomic analysis to neutrophils from patients with idiopathic pulmonary fibrosis (IPF), a non-infectious fibrotic lung disease. Additionally, immunofluorescence staining was performed on lung biopsy specimens from IPF patients to validate transcriptomic findings at the tissue level.
Results
Our analysis revealed significant transcriptional changes in COVID-19 neutrophils, particularly in pathways involved in immune regulation, inflammation, and antiviral responses. Additionally, pathways associated with autophagy and chromatin remodeling were upregulated, while translation-related processes were suppressed, indicating an increased predisposition for neutrophil extracellular trap (NET) release. This neutrophil transcriptional signature in COVID-19 appears to be associated with the previously reported deregulation of the Activin/Follistatin system in the periphery. Notably, a comparative transcriptomic analysis with neutrophils isolated from IPF patients revealed the induction of substantially overlapping inflammatory processes, suggesting common deregulated responses in COVID-19 and IPF. Consistently, significant NET formation, a hallmark of COVID-19-related inflammation, was observed within lung biopsies from IPF patients.
Conclusion
By delineating both shared and disease-specific molecular pathways, our findings validate the critical role of neutrophils in COVID-19 and IPF pathophysiology, highlighting their involvement in balancing the inflammatory response across diverse lung diseases.
Background
Coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has emerged as a global health crisis, with its severe impact largely attributed to respiratory complications, such as the acute respiratory distress syndrome (ARDS) and pulmonary fibrosis [1–2]. ARDS is marked by widespread lung inflammation, edema, and complex dysregulation of inflammatory and coagulation pathways [3], while pulmonary fibrosis involves aberrant extracellular matrix deposits within lung tissue that impair gas exchange, primarily driven by activated myofibroblasts [4], with both conditions leading to respiratory failure and potentially fatal outcomes. Neutrophils, the most abundant circulating white blood cells, are key mediators of the deregulated lung inflammatory response that drives COVID-19 pathophysiology [5].
As critical components of the innate immune system, neutrophils are rapidly recruited to sites of infection or injury, where they mediate both pathogen clearance and regulation of inflammatory responses [6]. Upon activation, neutrophils deploy a plethora of antimicrobial and pro-inflammatory molecules through degranulation and engage in phagocytosis to neutralize invading pathogens [6–7]. Additionally, they form neutrophil extracellular traps (NETs), which are web-like chromatin structures decorated with enzymes, such as neutrophil elastase and myeloperoxidase (MPO), that trap, inactivate, and kill pathogens [8,9,10]. However, while neutrophils are essential for host defense, their excessive or prolonged activation can lead to uncontrolled inflammation and collateral tissue damage, highlighting their dual responses in health and disease [11–12].
Increased neutrophil counts and an elevated neutrophil-to-lymphocyte ratio (NLR) have been strongly associated with COVID-19 severity, underscoring neutrophils’ contribution to the hyperinflammatory state observed in severe cases [13–14]. Excessive NET formation in COVID-19 has also been implicated in lung injury and poor clinical outcomes [15,16,17,18]. Despite substantial research into COVID-19 pathophysiology, the specific molecular pathways by which neutrophils drive inflammatory processes compromising lung function remain poorly understood, as much of the current knowledge is mainly based on bulk or single-cell transcriptomic analysis of whole blood samples [19,20,21].
To bridge this knowledge gap, we conducted a comprehensive transcriptomic analysis of neutrophils isolated from the peripheral blood of COVID-19 patients. Our findings revealed significant alterations in immune regulatory, inflammatory, and antiviral pathways, marked by a prominent interferon signature and an increased tendency for NET formation. This heightened inflammatory neutrophil state appears to be partially attributed to the deregulation of the Activin/Follistatin system, which has been previously linked to disease severity and higher in-hospital mortality in COVID-19 patients [22–23].
Further, to validate our findings, we compared the transcriptome of COVID-19 neutrophils with that of neutrophils isolated from patients with idiopathic pulmonary fibrosis (IPF), a non-viral chronic interstitial lung disease characterized by irreversible loss of lung function due to excessive extracellular matrix deposition and distortion of lung architecture [24–25]. Aberrant neutrophil activation has been implicated in IPF progression through the release of neutrophil elastase and other pro-inflammatory mediators that contribute to tissue remodeling [26,27,28,29]. Interestingly, neutrophils from IPF patients displayed a significant overrepresentation of inflammatory and NETosis-related pathways similar to COVID-19 neutrophils, underscoring their involvement in chronic inflammation and fibrosis.
Collectively, the overlapping transcriptomic profiles of COVID-19 and IPF neutrophils reveal robust transcriptional reprogramming that drives a shared neutrophil-driven mechanism underlying both viral and non-infectious lung injuries. This shared molecular signature, encompassing inflammatory and NETosis-related pathways, offers potential biomarkers for assessing neutrophil activation and promising therapeutic targets to reduce collateral tissue damage in respiratory diseases.
Methods
Patients
Peripheral blood samples were collected from eleven COVID-19 patients (Table S1), who were admitted to the ‘Attikon’ General University Hospital, Athens, Greece, during the initial phase of the pandemic, from April 7 to April 25, 2020. The diagnosis of SARS-CoV-2 infection was confirmed based on the following criteria: (i) the presence of a compatible clinical picture lasting between 5 and 14 days, and (ii) a positive real-time PCR test for SARS-CoV-2 RNA from a nasopharyngeal swab. All COVID-19 patients received standard-of-care treatment, as determined by the attending physician, which included low molecular weight heparin, antibiotic prophylaxis, and supportive care. Additionally, peripheral blood samples were collected from six patients with IPF (Table S2) who were admitted to the ‘Sotiria’ General Hospital of Chest Diseases, Athens, Greece. Lung biopsies from a separate cohort of IPF patients from the same hospital were also included in the study. In these patients, lung biopsy was deemed necessary to obtain histopathological data and confirm the diagnosis of IPF. The biopsies were obtained via video-assisted thoracoscopic surgery as part of the diagnostic workup, in accordance with the ATS/ERS/JRS/ALAT Guidelines 2018 [30]. Tables S1 and S2 provide key demographic, hematological, and disease-related parameters for COVID-19 and IPF patients, respectively. The control group consisted of eleven healthy individuals (HI) who had no history of COVID-19 or IPF at the time of inclusion.
Isolation of peripheral blood neutrophils
Peripheral blood was collected in heparin-coated tubes, and neutrophils were isolated by Histopaque (Sigma-Aldrich, 1077 and 1119) double-gradient density centrifugation [900×g, 35 min, room temperature (RT)], as previously described [31]. Isolated neutrophils were centrifuged (300×g, 10 min, RT), and the cell pellets were lysed in 1 ml TRI Reagent®.
RNA isolation and sequencing
RNA extraction was performed using the TRI Reagent® (Sigma-Aldrich, T9424), according to the manufacturer’s instructions. RNA quality was assessed using a NanoDrop spectrophotometer (ThermoFisher Scientific), with 260/280 absorbance ratios of approximately 2.0 obtained across all samples, indicating intact RNA. Electrophoretic analysis on 2% agarose gels (Sigma-Aldrich, A9539) further confirmed RNA integrity, as evidenced by the presence of sharp 28S and 18S rRNA bands. For cDNA library preparation, 1 μg of total RNA was processed using the TruSeq RNA Library Preparation Kit v2 (Illumina, RS-122-2002), as previously described [32–33]. Sequencing was carried out at the Greek Genome Center (BRFAA, Athens, Greece), where all cDNA libraries were sequenced simultaneously in the same run on a NovaSeq 6000 SP 100c platform (Illumina, 20028401), generating single-end 100 bp reads with adequate sequencing depth while minimizing potential batch effects.
Bioinformatics analysis
Raw sequence data were uploaded to the Galaxy web platform [34], and standard tools of the public server “usegalaxy.org” were used for subsequent analysis, as previously described [31–32]. In brief, HISAT2 (v2.2.1 + galaxy1) was applied for the alignment of trimmed reads to the human genome assembly GRCh37/hg19 (Genome Reference Consortium). Uniform read coverage was assessed using the Gene Body Coverage tool (v2.6.4.3) to exclude 5’/3’ bias, while RNA integrity was further confirmed at the transcript level with the Transcript Integrity Number tool (v2.6.4.1). The identification of differentially expressed genes (DEGs) between HI and patients with either COVID-19 or IPF was performed using the DESeq2 algorithm (v2.11.40.7 + galaxy2) [35]. Raw counts generated with the HTSeq-count tool (v0.9.1) were normalized using DESeq2, which applies variance-stabilizing transformation and corrects for differences in sequencing depth, ensuring robust and reliable log2 fold change calculations for all transcripts. To ascertain the purity of our samples, absolute deconvolution of human immune cell types was performed using the Shiny app, https://giannimonaco.shinyapps.io/ABIS [36]. This analysis ensured an enrichment of granulocyte gene expression signatures (exceeding 95%). Replicates displaying enrichment of contaminating lymphocyte signatures (greater than 5%) were excluded from further analysis.
The cutoff values used for statistically significant DEGs were baseMean > 30 and adjusted p-value (false discovery rate, FDR) < 0.05. These widely accepted thresholds provided a robust foundation for identifying reliable gene expression changes. Pathway analysis was subsequently performed using the GeneCodis4 web-based tool [37]. Gene set enrichment analysis (GSEA) was performed using the GSEA software (University of California, San Diego & Broad Institute, USA), as previously described [38–39]. Briefly, normalized counts generated with DESeq2 and annotated gene sets from the Human Molecular Signatures Database (Human MSigDB v2024.1) were used as inputs. Gene sets were ranked by taking the -log10 (p-value) and signed as positive or negative based on the direction of fold change, followed by pre-ranked analysis using the default settings (1000 permutations, min and max term size of 15 and 500, respectively). GSEA plots were annotated with key statistical metrics: normalized enrichment score (NES), p-value, and FDR. The NES quantifies the magnitude of enrichment and is normalized across gene sets to enable comparison between analyses, with positive values indicating pathway activation and negative values indicating pathway suppression.
Principal component analysis (PCA) plots were generated automatically using the DESeq2 tool on the Galaxy web platform. Volcano, bar, and violin plots were created with the GraphPad Prism software (v8.4.3, San Diego, CA, USA). Heatmaps were generated using the Morpheus software, https://software.broadinstitute.org/morpheus (Broad Institute, USA). Venn diagrams were constructed with Venny 2.1 (developed by Oliveros, J.C., 2007) and Venn Diagram Plotter software (Pacific Northwest National Laboratory, U.S. Department of Energy).
Immunofluorescence analysis
Immunostaining was performed on paraffin-embedded lung biopsy sections. Prior to immunostaining, tissue sections were deparaffinized in xylene and rehydrated through a series of graded ethanol dilutions, followed by immersion in dH2O. Following antigen retrieval in 10 mM sodium citrate buffer (pH 6.0) at 80°C for 30 min, tissues were blocked with 2% donkey serum (Sigma-Aldrich, D9663) in PBS containing 0.2% Triton-X (PBS-TX) for 60 min at RT. Sections were then incubated overnight at 4°C with rabbit anti-histone H3 (Abcam, ab1791, 1:200) and goat anti-MPO (R&D Systems, AF3667, 1:100) primary antibodies in PBS-TX. After washing three times with PBS, sections were treated for 60 min at RT in the dark with the following donkey-raised secondary antibodies in PBS-TX: anti-rabbit Alexa 488 (Jackson ImmunoResearch, 711-546-152, 1:300) and anti-goat Alexa 647 (Jackson ImmunoResearch, 705-605-147, 1:300). DNA staining, including nuclear and extracellular chromatin, was performed using 4′,6-diamidino-2-phenylindole (DAPI) dihydrochloride (Calbiochem, 268298, 2.5 μg/ml). Sections were mounted with a fluorescence mounting medium (Dako, S3023). Images were captured using a Leica TCS-SP5II confocal microscope (Leica Microsystems, Germany) and analyzed with the Fiji/ImageJ v2.0.0 software.
Results
Transcriptomic analysis reveals neutrophil hyperactivation in COVID-19 patients
To uncover the transcriptional changes underlying neutrophil-driven pathology in COVID-19, we performed a comprehensive transcriptomic analysis of neutrophils isolated from the peripheral blood of SARS-CoV-2-infected patients. Principal component analysis (PCA) revealed distinct clustering between neutrophils from COVID-19 patients and HI, indicating significantly different transcriptional profiles (Fig. 1A). A total of 2536 transcripts were upregulated, and 1937 were downregulated in COVID-19 patient neutrophils compared to the control group (Fig. 1B, Table S3). Table S3 provides the full list of these DEGs, along with their respective fold changes and statistical values. Pathway analysis using the GeneCodis4 web-based tool revealed the upregulation of key immune-regulatory, inflammatory, and antiviral pathways (Fig. 1C). Notably, inflammatory pathways, including defense response to virus, cytokine signaling, interferon signaling, Toll-like receptor (TLR) cascades, positive regulation of NF-κB transcription factor activity, and class I MHC-mediated antigen processing and presentation, were significantly enriched, indicating robust activation of innate immunity in response to SARS-CoV-2 infection. Additionally, upregulation of autophagy-related pathways, signaling by Rho GTPases, chromatin organization and remodeling components, including chromatin-modifying enzymes, as well as neutrophil degranulation (Fig. 1C), highlighted the pivotal role of neutrophils in viral recognition and aligned with a NETosis-prone molecular signature, consistent with previous reports of increased NET formation during COVID-19 [15–16, 20, 40]. Moreover, processes related to gene expression, including both positive and negative regulation of transcription by RNA polymerase II, were overrepresented, as were pathways related to protein ubiquitination, phosphorylation, vesicle-mediated transport, and protein trafficking, reflecting extensive post-translational modifications and intracellular trafficking during SARS-CoV-2 infection.
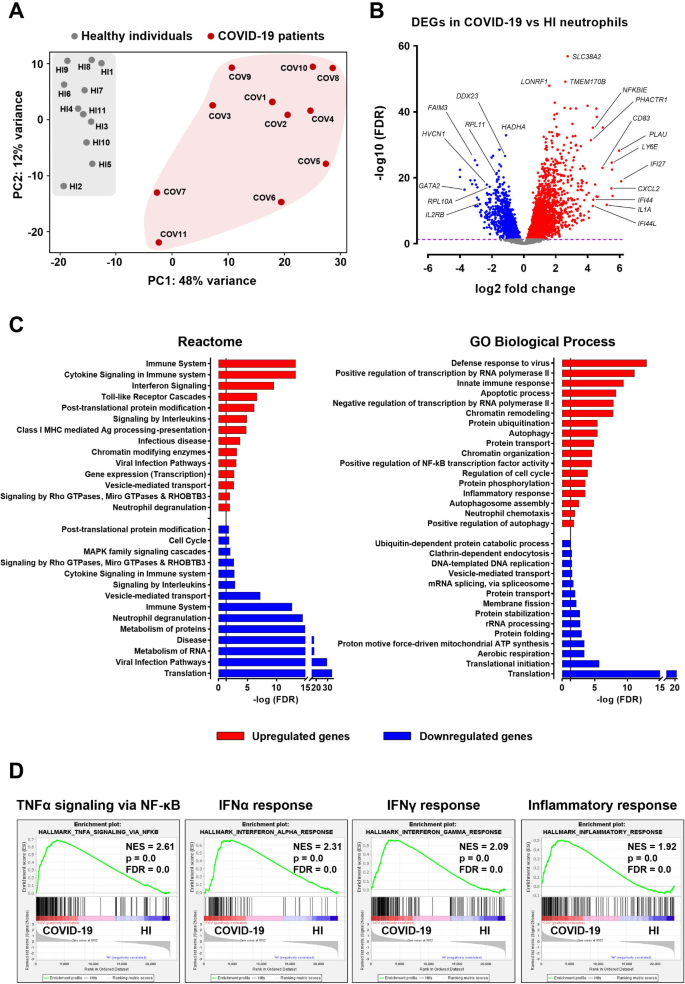
Conversely, neutrophils from COVID-19 patients exhibited marked downregulation in pathways linked to protein synthesis and metabolic processes (Fig. 1C). More specifically, translation, translational initiation, protein folding and stabilization, and rRNA processing were significantly suppressed, indicating a reduced capacity for protein production and degradation. Metabolic processes, including aerobic respiration, mitochondrial ATP synthesis, and metabolism of RNA and proteins, were also underrepresented. Moreover, downregulation of processes such as clathrin-dependent endocytosis, membrane fission, cell cycle, and MAPK family signaling cascades further underscores neutrophils’ shift from homeostatic and regulatory roles towards a highly inflammatory state during SARS-CoV-2 infection (Fig. 1C). Interestingly, we observed that several immune-related pathways, such as neutrophil degranulation, interleukin signaling, and cytokine signaling, included both upregulated and downregulated genes, suggesting a potential dysregulation or exhaustion of immune responses in COVID-19 patients.
Gene set enrichment analysis (GSEA) using the Hallmark gene set collection of the Human Molecular Signatures Database further validated the inflammatory hyperactivation of COVID-19 neutrophils, identifying TNFα signaling via NF-κΒ, inflammatory response, as well as IFNα and IFNγ responses, among the most enriched molecular signatures (Fig. 1D).
Correlation of neutrophil transcriptional alterations with the Activin/Follistatin axis and disease markers in COVID-19
We and others have previously demonstrated deregulation of the Activin/Follistatin system during COVID-19, where elevated serum levels of Activin-A, Activin-B, and Follistatin correlate with disease severity and in-hospital mortality [22–23]. Building on these findings, we analyzed the relationship between the transcriptional profile of COVID-19 neutrophils and serum levels of Activins and Follistatin, along with classic neutrophil-related disease markers such as absolute neutrophil count (ANC) and neutrophil-to-lymphocyte ratio (NLR). We focused on the most prominently altered pathways in COVID-19 neutrophils, namely interferon signaling, TLR cascades, autophagy-related processes, and translation (Fig. 2A), and calculated Pearson correlation coefficients between DEGs involved in these pathways and patient serum biomarkers.
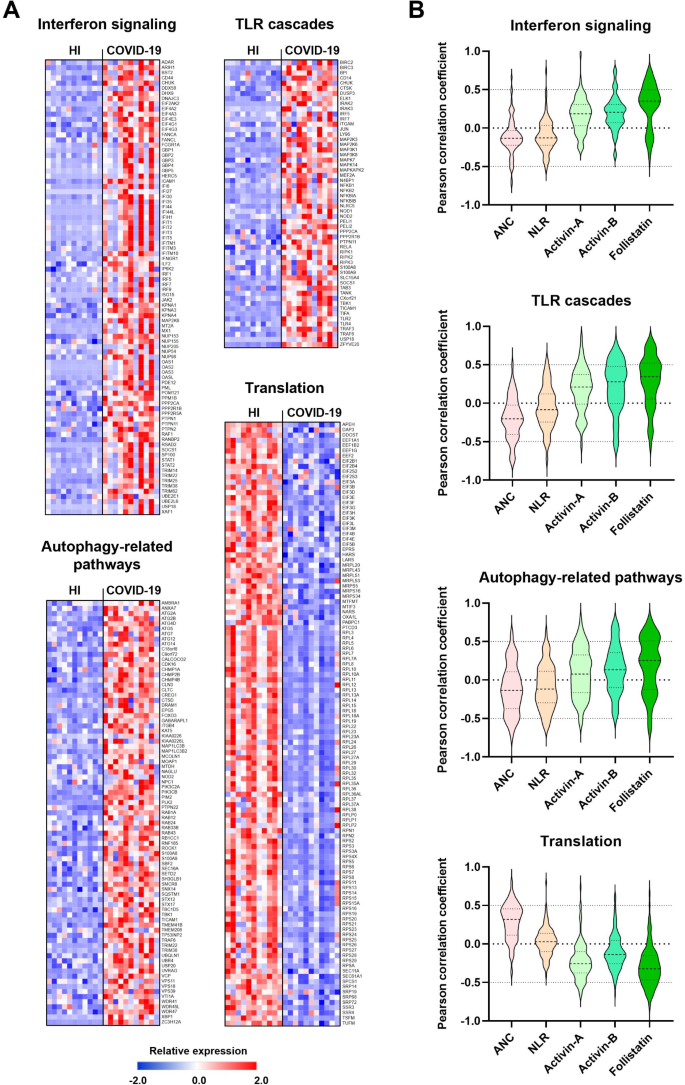
Interestingly, our analysis revealed that serum levels of Activins and Follistatin positively correlated with DEGs in the upregulated pathways, namely interferon signaling, TLR cascades, and autophagy-related processes, while no significant correlation emerged between ANC or NLR and these same pathways (Fig. 2B). In contrast, the downregulated pathway of translation was negatively correlated with Activins and Follistatin but showed a positive correlation with ANC, with no significant association with NLR (Fig. 2B). Across these pathways, Follistatin consistently showed a tendency for stronger correlations with altered gene expression than Activins. These findings indicate that deregulation of the Activin/Follistatin axis in COVID-19 patients positively correlates with the transcriptional reprogramming observed in these neutrophils, potentially influencing critical inflammatory and immune-related responses.
Transcriptomic profiling of neutrophils in IPF patients identifies an inflammatory signature with pro-NETotic characteristics
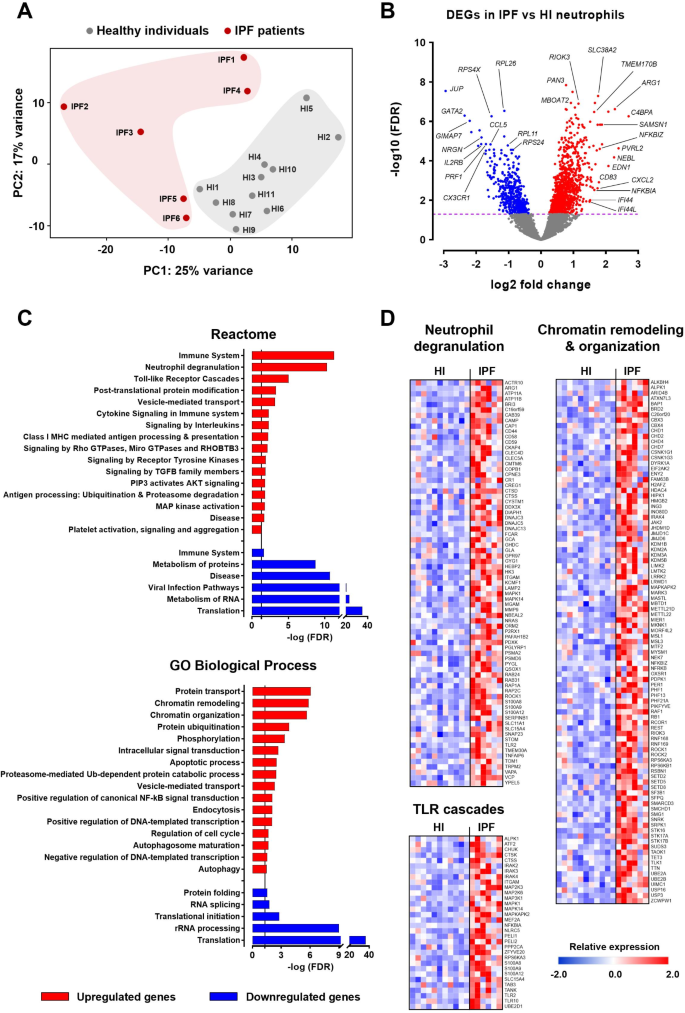
To validate the neutrophil signature observed in COVID-19 and assess its specificity to this viral infection, we sought to analyze the neutrophil transcriptome of patients suffering from a non-infectious inflammatory lung disease. We thereby analyzed the transcriptome of neutrophils isolated from the peripheral blood of patients with IPF, a progressive fibrotic lung disease marked by worsening respiratory symptoms, extensive tissue remodeling, and abnormal neutrophil activity [26,27,28,29]. Principal component analysis of the transcriptomic data revealed distinct clustering of neutrophils from HI and IPF patients (Fig. 3A). Differential expression analysis identified 1127 upregulated and 491 downregulated genes in IPF neutrophils compared to healthy controls (Fig. 3B, Table S4). Table S4 provides the full list of these DEGs, along with their respective fold changes and statistical values. Pathway analysis using GeneCodis4 revealed significant upregulation of inflammatory and immune response pathways, including neutrophil degranulation, TLR cascades, cytokine signaling in immune system, signaling by interleukins, class I MHC-mediated antigen processing and presentation, apoptotic process, and positive regulation of canonical NF-κB signal transduction (Fig. 3C-D). Of note, increased neutrophil degranulation, alongside additional upregulated processes such as chromatin organization and remodeling, autophagy, autophagosome maturation, and signaling by Rho GTPases, were suggestive of a heightened predisposition for NET release. The transcriptional reprogramming observed in IPF neutrophils also included upregulation of genes related to positive and negative DNA-templated transcription, protein transport and ubiquitination, phosphorylation, as well as various signaling mechanisms mediated by transforming growth factor beta (TGFβ) family members, AKT and mitogen-activated protein (MAP) kinases, and receptor tyrosine kinases (Fig. 3C). This aligns with previous reports of genome-wide transcriptional activation regulated by specific kinase cascades, including MAP and AKT kinases, which are essential for chromatin decondensation and subsequent NET formation [41]. In contrast, several pathways were downregulated, including those involved in translation and the metabolism of RNA and proteins (Fig. 3C).
Identification of regions with prominent NET formation in lung biopsies from IPF patients
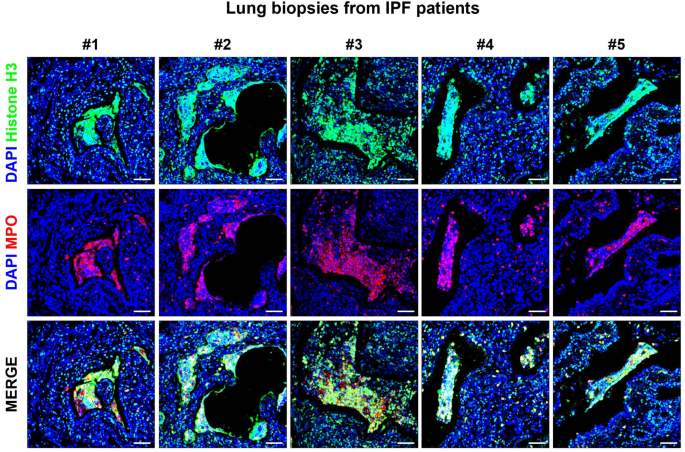
Given that several altered pathways in IPF neutrophils, such as increased neutrophil degranulation, autophagy, and chromatin remodeling, together with the downregulation of translation-related processes, indicated an increased pro-NETotic activity, we sought to investigate potential NET formation at the tissue level. To this end, we performed immunofluorescence staining on lung biopsy sections from IPF patients, using antibodies against histone H3 and MPO, two key markers of NETs.
We observed notable colocalization of histone H3 and MPO in areas with extensive extracellular chromatin deposition, indicating significant NET formation within the lung parenchyma and airspaces of IPF patients (Figs. 4, and S1). Importantly, NET formation was observed in several areas of the lung tissue, although not homogeneously distributed, displaying interindividual and spatial heterogeneity. Similarly, previous studies have identified NETs in postmortem lung specimens from COVID-19 patients [42–43]. The presence of these structures within tissue samples not only validates our transcriptomic findings but also reinforces the contribution of NETosis in both COVID-19 and IPF pathophysiology, underlying the relevance of NETs as potential contributors to disease progression.
Comparative transcriptomic analysis of neutrophils in COVID-19 and IPF reveals shared and disease-specific pathways
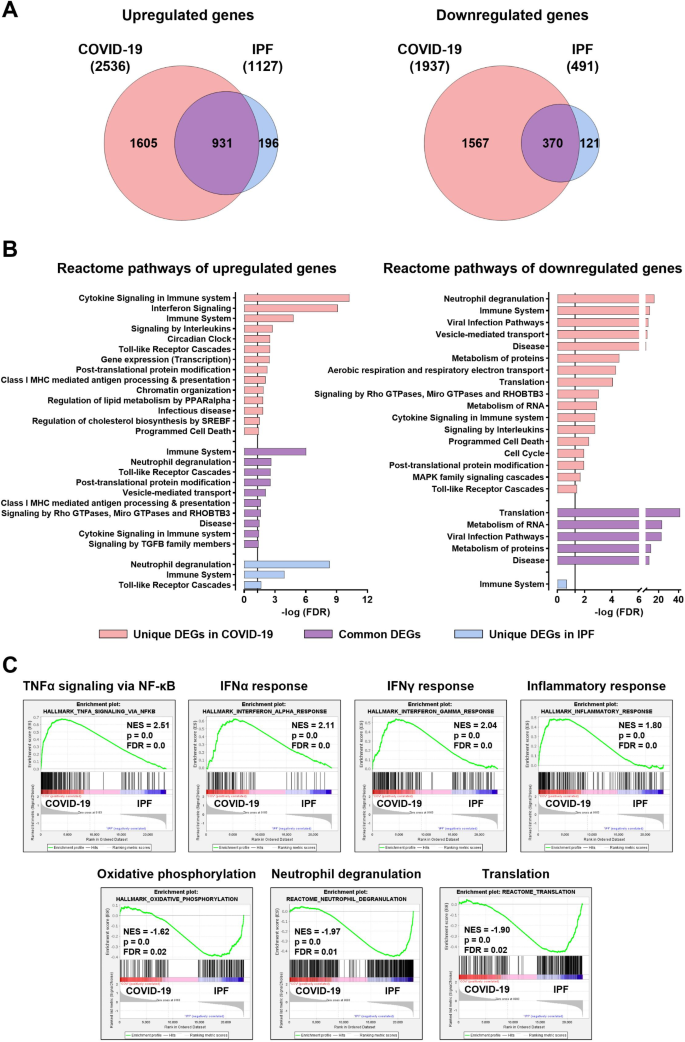
Next, we performed a comparative analysis of neutrophil transcriptomes from COVID-19 and IPF patients. Surprisingly, these transcriptomes showed remarkable overlap, with 82% of upregulated (n = 931) and 75% of downregulated (n = 370) genes in IPF neutrophils being also differentially expressed in COVID-19 neutrophils (Fig. 5A, Table S5). Table S5 details the common and unique DEGs in neutrophils from both diseases. These findings may suggest a shared neutrophil response in inflammatory lung diseases, regardless of whether they are of infectious or non-infectious origin. Pathway analysis indicated that commonly upregulated genes primarily clustered in immune response pathways, including neutrophil degranulation, TLR cascades, class I MHC-mediated antigen processing and presentation, cytokine signaling in immune system, as well as signaling by TGFβ family members (Figs. 5B and 6A-D, and S2). Autophagy, chromatin remodeling, and signaling by Rho GTPases were also upregulated, indicative of a heightened pro-NETotic tendency (Figs. 5B and 6E-G, and S2). Moreover, vesicle-mediated transport, post-translational protein modification, and protein ubiquitination were also commonly enriched, further supporting the pro-inflammatory status of activated neutrophils (Figs. 5B and 6H, and S2). Shared downregulated pathways included translation-related processes, metabolism of RNA and proteins, cellular defense response, and calcium-mediated signaling (Figs. 5B and 6I-J, and S2).

Further, to highlight neutrophil-associated DEGs and pathways linked to lung inflammation, we conducted a comparative transcriptomic analysis between the shared inflammatory signature identified herein for lung diseases and the recently characterized neutrophil transcriptome in inflammatory bowel disease [31], which includes the chronic neutrophil-related intestinal disorders Crohn’s disease and ulcerative colitis. This analysis revealed a limited overlap between the inflammatory signatures in lung and intestinal diseases, with no more than 42% of DEGs shared in any comparison (Fig. S3A, Table S6). Table S6 outlines the common and unique DEGs in neutrophils from patients with inflammatory lung and bowel diseases, thus highlighting their distinct molecular signatures. Notably, 462 genes were uniquely upregulated and 187 uniquely downregulated in inflammatory lung diseases (Fig. 6, Table S6). The uniquely upregulated genes prominently clustered into key pathways previously analyzed, including chromatin organization and remodeling, protein transport and ubiquitination, regulation of gene expression, and TLR signaling (Fig. S3B). In contrast, the uniquely downregulated genes were mainly associated with translation and RNA processing (Fig. S3B). These findings suggest a unique transcriptional landscape in neutrophils during lung inflammation, characterized by concurrent activation of inflammatory signaling pathways and suppression of the translational machinery, potentially contributing to the neutrophil-driven pathology observed in both COVID-19 and IPF.
Beyond shared DEGs, several genes and pathways were uniquely regulated in each disease. Notably, unique DEGs in COVID-19 significantly outnumbered those in IPF, with 1605 unique upregulated and 1567 unique downregulated genes, compared to 196 and 121, respectively, in IPF (Fig. 5A, Table S5). Unique upregulated genes in COVID-19 clustered in pathways related to immune response, such as interferon signaling, signaling by interleukins, and TLR cascades, reflecting the SARS-CoV-2 ability to elicit a robust inflammatory response (Figs. 5B, S2, and S4A). Additional unique upregulated pathways, including transcription, post-translational protein modification, chromatin organization, regulation of lipid metabolism by PPARα, regulation of cholesterol biosynthesis by SREBF, and programmed cell death, indicate a complex interplay between immune activation and metabolic reprogramming in response to viral infection. COVID-19-specific downregulated pathways encompassed neutrophil degranulation, viral infection pathways, vesicle-mediated transport, metabolism of proteins and RNA, aerobic respiration, translation, and post-translational protein modification (Figs. 5B, S2, and S4A), indicating potential impairment in response to pathogens, coupled with decreased metabolic and translational activities during inflammation. Additional uniquely downregulated pathways in COVID-19 neutrophils included interleukin signaling, programmed cell death, cell cycle, and MAPK family signaling.
Conversely, unique upregulated genes in IPF neutrophils primarily clustered in neutrophil degranulation, TLR cascades, and immune system pathways (Figs. 5B, S2, and S4B), underscoring the role of neutrophil-driven inflammation in promoting fibrotic processes in this chronic disease. Finally, GSEA analysis indicated distinct molecular signatures for each disease, regardless of comparisons with healthy individuals. In COVID-19 neutrophils, enriched signatures included TNFα signaling via NF-κB, IFNα and IFNγ responses, and inflammatory response, whereas IPF neutrophils were characterized by increased neutrophil degranulation, oxidative phosphorylation, and translation signatures (Fig. 5C).
Overall, although unique molecular pathways were identified in COVID-19 and IPF neutrophils, both share a prominent common inflammatory signature (Fig. 6). This shared signature could serve as a hallmark for neutrophil activation in inflammatory lung diseases of irrespective etiology and may be employed as a therapeutic target to tame neutrophil overactivation in relevant pathological conditions.
Discussion
In COVID-19, increased neutrophil-related laboratory parameters, such as ANC and NLR, as well as extensive NET formation, are closely associated with disease severity, highlighting the role of neutrophils in driving a hyperinflammatory response that compromises lung function [13–14, 17]. However, the precise molecular mechanisms by which neutrophils mediate viral clearance and contribute to disease pathology remain elusive. This study offers an extensive transcriptomic analysis of peripheral blood neutrophils from COVID-19 patients, revealing significant gene expression changes that drive hyperactivation of inflammatory pathways. Moreover, by comparing the neutrophil transcriptome in COVID-19 to that in IPF–an inflammatory interstitial lung disease of non-viral origin [24–25] – we pinpoint both shared and disease-specific pathways, offering new insights into potential targets for mitigating neutrophil-mediated damage in pathological lung conditions.
Our study demonstrated substantial transcriptional reprogramming of peripheral blood neutrophils in response to SARS-CoV-2 infection, with over 4000 transcripts significantly altered compared to healthy individuals. We identified a shift from homeostatic functions to an inflammatory state, characterized by the overrepresentation of immune-regulatory pathways, including interferon signaling, TLR cascades, and NF-κB activation, all essential for initiating and propagating antiviral responses. This aligns with our prior findings demonstrating deregulation of interferon responses in COVID-19, coupled with the early and sustained production of pro-inflammatory cytokines like TNFα, IL-6, and IL-8, contributing to untuned antiviral responses, prolonged inflammation, and heightened risk of respiratory failure [44]. Moreover, while preparing this manuscript, emerging transcriptomic analysis of polymorphonuclear cells demonstrated a significant type I interferon gene signature in patients with severe COVID-19, in contrast to those with mild COVID-19 and healthy individuals [45]. These are consistent with findings from whole blood transcriptome studies, which highlight granulocytes as central contributors to COVID-19 severity [20], and from integrated single-cell RNA-Seq data and genome-wide association studies that demonstrate a significant association between neutrophils and SARS-CoV-2 infection within a European cohort [46].
In addition to the COVID-19 dataset, our study is the first to present a transcriptomic analysis of neutrophils in IPF patients, revealing significant transcriptional changes in comparison to control neutrophils. Notably, more than 80% of DEGs in IPF were similarly regulated in COVID-19 neutrophils, with inflammation-related pathways such as neutrophil degranulation and TLR cascades overrepresented, suggesting an intensified pro-inflammatory response in both diseases. These observations are consistent with prior studies reporting increased release of pro-inflammatory mediators, such as neutrophil elastase, in the lung parenchyma, bronchoalveolar lavage (BAL), and serum [26, 28], alongside elevated neutrophil counts in blood and BAL samples, both linked to lung function decline and early mortality in IPF patients [27, 47]. Experimental data from murine models further suggest that enzymes released by neutrophils may exacerbate tissue damage and fibrosis through TGFβ pathway activation and fibroblast proliferation [29].
The overlap in inflammatory pathways between COVID-19 and IPF, despite their distinct etiologies, reflects the fundamental role of neutrophils as frontline responders to tissue injury. In COVID-19, viral RNA activates TLRs, driving excessive type I interferon production, pro-inflammatory cytokine release, and neutrophil activation [48–49]. Likewise, in IPF, persistent epithelial damage and abnormal wound healing processes activate sterile inflammation through similar TLR-dependent mechanisms [50–51]. Additionally, common cytokines, chemokines, or growth factors released in the inflamed lung tissue by resident lung cells or infiltrating immune cells may further amplify neutrophil activation in both diseases.
Furthermore, the upregulation of autophagy-related pathways, neutrophil degranulation, and chromatin organization and remodeling components, coupled with suppressed protein synthesis and metabolic-related processes, observed in both COVID-19 and IPF, suggests that activated neutrophils undergo functional reprogramming toward a pro-NETotic phenotype [52,53,54,55]. Additionally, the prominentcolocalization of histone H3 and MPO observed in areas with extensive extracellular chromatin deposition in lung biopsies, indicative of significant NET formation in the lung parenchyma and airspaces of IPF patients, reinforces our transcriptomic findings. These observations align with previous reports of increased NET formation in COVID-19 [15–16, 20, 40, 42–43], interstitial lung disease [56–57], and more recently in IPF [58], highlighting the role of NETosis-related mechanisms in the immune response to SARS-CoV-2 and their potential contribution to profibrotic processes in chronic lung diseases. In line with this, we have recently reported that activated neutrophils can acquire an immunofibrotic role through NET release in Crohn’s disease [31], suggesting that activated neutrophils may similarly trigger profibrotic cascades in both COVID-19 and IPF through interaction with myofibroblasts.
Building on these findings, the relationship between transcriptional changes in neutrophils and NET formation remains unclear–are these changes a cause or consequence of NETosis? While studies such as Khan and Palaniyar’s suggest that transcriptional firing facilitates NETosis by promoting chromatin decondensation and regulating distinct gene sets [41], contrasting evidence, like that from Sollberger et al., indicates that transcription may not be essential in certain contexts [54]. This discrepancy points to a context-dependent role of transcription in NETosis, varying with the disease-associated inflammatory environment, specific stimuli, or pathways involved. Additionally, NETs along with their associated enzymes and proteins may act as paracrine signals that target neighboring neutrophils, altering their transcriptome and potentially creating a feedback loop that amplifies inflammation and promotes neutrophil hyperactivation. Therefore, further research is needed to dissect these mechanisms and clarify whether transcriptional alterations actively drive NETosis or emerge as a secondary consequence of the process.
Overall, given their potential to drive pathology in inflammatory lung conditions, NETs present as promising therapeutic targets to modulate disease progression. Thus, current antifibrotic therapies [59–60] could be combined with agents targeting NETosis and other inflammatory pathways, offering a complementary approach against neutrophil hyperactivation. Potential interventions include DNase I to target NET formation, neutrophil elastase inhibitors (e.g. alvelestat), modulators of TGFβ signaling (e.g. galunisertib), oligonucleotide-based TLR antagonists, and Janus kinase inhibitors [61,62,63]. For instance, the successful inhalational administration of human recombinant DNase I (Pulmozyme/dornase alfa) in cystic fibrosis [64–65] and COVID-19-associated ARDS [66,67,68] may offer potential benefits in treating IPF as well.
Pulmonary fibrosis is a severe complication in COVID-19 and a frequently reported post-infectious lung condition that significantly impacts patient outcomes [2]. Emerging data indicate that clinical risk factors for severe COVID-19 and post-COVID pulmonary fibrosis–such as advanced age, male sex, and comorbidities like hypertension and diabetes – overlap with those of IPF patients, suggesting that COVID-19 and IPF may share common pathogenetic pathways [69]. Additionally, single-cell RNA-Seq data from lung tissue of severe COVID-19 patients revealed fibrogenic gene signatures similar to those observed in IPF, indicating that comparable gene expression patterns, particularly involving profibrogenic genes, contribute to pulmonary fibrosis regardless of the underlying cause [70]. This highlights the potential of common pathways as therapeutic targets and the need for developing biomarkers to guide precision medicine. Biomarkers–particularly those easily identified in blood samples – could enable patient stratification based on their inflammatory profiles, facilitating personalized therapies that integrate antifibrotic and anti-inflammatory approaches to improve disease outcomes.
Of interest, in COVID-19 neutrophils, the interferon signature is accompanied by reduced activity in neutrophil degranulation and oxidative phosphorylation pathways, in contrast to IPF neutrophils. This may reflect a state of neutrophil exhaustion, where hyperactivated neutrophils lose effectiveness in viral clearance, potentially exacerbating tissue damage in COVID-19. These findings are consistent with previous studies demonstrating that hyperactivated neutrophils in critically ill COVID-19 patients exhibit lower L-selectin expression and diminished oxidative burst responses, contributing to an increased risk of secondary infections [71].
The Activin/Follistatin axis, a branch of the TGFβ superfamily system, has been reportedly deregulated in COVID-19, with elevated serum levels of Activin-A, Activin-B, and particularly Follistatin closely linked to disease severity and in-hospital mortality [22–23]. Among these, Follistatin, the natural inhibitor of Activin-A, is the most significant predictor of poor outcomes across all disease stages [23]. Our findings demonstrate a significant positive correlation between neutrophil transcriptional changes and the deregulated Activin/Follistatin system in COVID-19, with high levels of Activins and Follistatin linked to DEGs involved in inflammation and immune response pathways, including interferon signaling, TLR cascades, and autophagy. Recent studies from our group further support these observations, linking Activin-A deficiency to a pro-inflammatory and NETosis-prone neutrophil phenotype in a murine model of viral infection [39]. Additionally, upregulation of key TGFβ superfamily signaling components, such as the receptors ACVR1B/ALK4 and TGFBR1/ALK5, and the intracellular mediator SMAD4, in both COVID-19 and IPF neutrophils further emphasizes the role of the TGFβ superfamily in driving the heightened neutrophilia that characterizes these lung diseases.
These findings highlight the translational potential of the Activin/Follistatin axis as both a diagnostic and therapeutic target in neutrophil-driven inflammatory conditions. Our previously developed FACT-CLINYCoD scoring system, which integrates Activins, Follistatin, and other clinically relevant parameters, has shown promise in identifying high-risk patients, predicting outcomes, and dynamically monitoring COVID-19 progression [23]. This scoring system could also be adapted for use in other clinical contexts marked by abnormal neutrophil activation, thereby expanding its applicability beyond COVID-19.
Unexpectedly, ANC showed a negative correlation only with translation, while NLR did not correlate significantly with any of the identified pathways in COVID-19 neutrophils, suggesting that serum markers like Activins and Follistatin may be more indicative of neutrophil-related immune deregulation than the absolute number of neutrophils and lymphocytes per se. The availability of commercial kits for measuring Activin and Follistatin levels can facilitate their integration into routine diagnostic workflows, enhancing disease monitoring, guiding treatment strategies, and identifying patients at risk of adverse outcomes in various inflammatory conditions, including IPF. Moreover, therapeutic strategies aimed at restoring balance within the Activin/Follistatin axis or targeting Activin receptors may help mitigate neutrophil-driven hyperinflammatory conditions, ultimately reducing morbidity and mortality.
Overall, our study suggests that neutrophils may initiate a potent inflammatory transcriptional program during lung inflammation and remodeling, irrespective of the underlying etiology. Interestingly, over 50% of the identified DEGs appear to be uniquely regulated in inflammatory lung diseases, as revealed by a comparative transcriptomic analysis of neutrophils between inflammatory intestinal diseases, analyzed in [31], and the lung diseases presented in this study. Given that neutrophil overactivation is linked to pathology [11–12, 14], this core neutrophil signature could serve as a useful metric for assessing their activation during lung inflammation and as a potential source of therapeutic targets for inflammatory respiratory conditions.
Despite its novel findings, our study has several limitations that warrant consideration. First, the relatively small sample size, particularly for IPF patients, limits the generalizability of our conclusions. The inherent difficulty in obtaining samples from IPF patients emphasizes the necessity for larger and more diverse cohorts to validate our findings across various populations. Nevertheless, by focusing on high-quality samples and robust statistical thresholds, we aimed to maximize the reliability of our data within the constraints of limited sample availability.
Another key limitation is the reliance on peripheral blood neutrophils rather than tissue-resident neutrophils. While peripheral neutrophils provide insights into systemic immune activation, tissue-infiltrating neutrophils may exhibit distinct transcriptional signatures influenced by the inflamed lung microenvironment. Nevertheless, studies employing single-cell and spatial transcriptomic approaches, particularly in COVID-19 lungs, have shown that critical molecular pathways, including neutrophil degranulation, interferon and cytokine signaling, and inflammatory responses, are consistently enriched across both peripheral and tissue-resident neutrophils [72,73,74,75]. A similar pattern of transcriptional consistency in key molecular signatures between peripheral and tissue-infiltrating neutrophils has also been reported recently in other inflammatory diseases, such as Crohn’s disease [31]. These findings suggest that peripheral neutrophils may retain key molecular signatures upon tissue infiltration, supporting their role as surrogates for studying neutrophil-driven immune responses in inflamed tissues.
Moreover, the cross-sectional design of our study limits temporal insights into the dynamics of neutrophil activation during disease progression. Longitudinal sampling of both peripheral and tissue-resident neutrophils could elucidate how their transcriptomic profiles evolve over time, especially following therapeutic interventions or resolution of acute inflammation. Of course, validation in other lung inflammatory diseases of both viral (e.g. influenza virus infection) and non-infectious (e.g. chronic obstructive pulmonary disease, bronchiectasis) etiology, where neutrophils play a key role in the underlying pathology [76–77], would further strengthen our findings. Future studies addressing these limitations will enhance the robustness and translational relevance of our results, ultimately helping the identification of therapeutic targets to mitigate neutrophil-driven pathology in inflammatory lung diseases.
Conclusions
This study uncovers extensive transcriptional reprogramming toward a pro-NETotic phenotype in neutrophils during COVID-19, which significantly overlaps with the inflammatory patterns identified herein in IPF neutrophils. These findings suggest that similar neutrophil-driven inflammatory responses may contribute to the pathogenesis of both diseases. Importantly, transcriptional changes in COVID-19 neutrophils are positively correlated with the aberrant activation of the Activin/Follistatin system in the periphery, which reflects disease severity and outcome.
These insights into neutrophil-mediated responses during respiratory inflammatory conditions provide a foundation for developing targeted therapeutic strategies. Modulating neutrophil activity and mitigating their inflammatory responses, particularly through the regulation of NETosis, could provide effective treatments for COVID-19, IPF, and other inflammatory lung diseases. Additionally, further investigation of the Activin/Follistatin system and neutrophil-related biomarkers could advance precision medicine, allowing for more personalized approaches to improve patient outcomes.