Yixin Huang, Tongyun Wang, Lijie Zhong, et. al., Nature
Abstract
Coronaviruses remodel the intracellular host membranes during replication, forming double-membrane vesicles (DMVs) to accommodate viral RNA synthesis and modifications1,2. SARS-CoV-2 non-structural protein 3 (nsp3) and nsp4 are the minimal viral components required to induce DMV formation and to form a double-membrane-spanning pore, essential for the transport of newly synthesized viral RNAs3,4,5. The mechanism of DMV pore complex formation remains unknown. Here we describe the molecular architecture of the SARS-CoV-2 nsp3–nsp4 pore complex, as resolved by cryogenic electron tomography and sub tomogram averaging in isolated DMVs. The structures uncover an unexpected stoichiometry and topology of the nsp3–nsp4 pore complex comprising 12 copies each of nsp3 and nsp4, organized in 4 concentric stacking hexamer rings, mimicking a miniature nuclear pore complex. The transmembrane domains are interdigitated to create a high local curvature at the double-membrane junction, coupling double-membrane reorganization with pore formation. The ectodomains form extensive contacts in a pseudo-12-fold symmetry, belting the pore complex from the intermembrane space. A central positively charged ring of arginine residues coordinates the putative RNA translocation, essential for virus replication. Our work establishes a framework for understanding DMV pore formation and RNA translocation, providing a structural basis for the development of new antiviral strategies to combat coronavirus infection.
Main
Positive-stranded RNA viruses substantially remodel intracellular membranes during RNA replication, which is a common feature observed in picornaviruses, flaviviruses, noroviruses and coronaviruses1. The replication of coronaviruses, such as MERS-CoV, SARS-CoV-2 and mouse hepatitis virus (MHV), leads to the formation of DMVs in host cells to accommodate viral RNA synthesis and modifications6. Similarly, alphaviruses (such as chikungunya virus) and nodaviruses induce membrane spherules as replication organelles for viral genome replication, forming a large ring complex at the spherule neck7,8. Coronaviruses have a relatively large RNA genome and more than two-thirds (about 20 kb) of it encodes 16 non-structural proteins, together with host proteins, to constitute the coronavirus replication–transcription complex2. The DMVs provide a central hub for viral genomic RNA and mRNA synthesis and processing by localizing and concentrating the necessary factors such as the RNA-dependent RNA polymerase complex, protecting viral RNAs from the host cell surveillance system1,9.
Although the functional importance of DMVs in coronavirus replication is well established1, the mechanism by which DMVs form to scaffold the replication–transcription complex and to facilitate the translocation of newly synthesized viral RNA molecules for viral assembly and protein synthesis remains unclear. Recent studies using in situ cryogenic electron tomography (cryo-ET) identified a pore complex on DMVs in MHV- and SARS-CoV-2-infected cells3,4,5. This pore complex, with an estimated mass of around 3 MDa, was found to exhibit a six-fold-symmetry double-membrane-spanning architecture7,9. Ectopic expression of nsp3 and nsp4 induces DMV formation, and results in similar pore complexes on DMVs to those observed in coronavirus-infected cells10,11,12, revealing that nsp3 and nsp4 are the minimal viral components required to constitute the DMV pore complex. However, the resolution of these pore complexes resolved in situ is limited (about 20 Å)3,13. Thus, the molecular architecture of the DMV pore complex and the mechanistic role of nsp3 and nsp4 in pore formation remain elusive.
Here we describe the structure of the coronavirus DMV pore complex using isolated DMVs formed by the minimal viral components (nsp3 and nsp4) in vitro. By cryo-ET and subtomogram averaging, we have resolved the molecular architecture of the SARS-CoV-2 nsp3–nsp4 pore complex at an overall resolution of 4.2 Å. Our structure reveals an unexpected stoichiometry of the coronavirus DMV pore complex constituted by 12 copies each of nsp3 and nsp4, whereas the complex was identified to exhibit a six-fold symmetry and six copies of nsp3 were proposed to form the crown7. We show that the complex is a putative RNA translocation pore coordinated by a central positively charged arginine ring, essential to virus replication. The structure of our minimal coronavirus DMV pore complex suggests a mechanism of RNA translocation, paving new paths for future drug design targeting DMV pore formation and RNA transport during coronavirus infections.
Architecture of nsp3–nsp4 pore complex on DMV
Co-expression of coronavirus nsp3 and nsp4 is sufficient to induce the DMV pore formation10,11,14 and form a minimal DMV pore complex13. We therefore expressed the SARS-CoV-2 nsp3–nsp4 tandem polypeptide in HEK293F cells, which is proteolytically processed into nsp3 and nsp4 by the papain-like protease (PLpro) in nsp3, to generate DMVs. We engineered a TwinStrep-GFP tag in the amino terminus of nsp3 and purified the DMVs by streptavidin affinity chromatography. The existence of nsp3 and nsp4 in the DMV was confirmed by western blotting and mass spectrometry (Extended Data Fig. 1a–c). We then imaged the resulting vesicles containing nsp3–nsp4 and revealed a mixture of DMVs and other single-membrane vesicles (Fig. 1a and Extended Data Fig. 1d). The isolated DMVs tend to aggregate together and exhibit a characteristic double-membrane appearance, with the postulated nsp3–nsp4 pore complex connecting two membranes (Fig. 1a and Extended Data Fig. 1d,e). To resolve the composition and architecture of these nsp3–nsp4 pore complexes on DMVs, we collected a large cryo-ET dataset of DMVs and carried out sub tomogram averaging (Methods and Extended Data Table 1). We identified the nsp3–nsp4 pore complex by template-matching from the reconstructed tomograms15 and carried out extensive three-dimensional (3D) classifications (Methods and Extended Data Fig. 2). Sub tomogram averaging with six-fold symmetry revealed three main populations of pore complex varying in the height of the cytoplasmic regions: most of the pore containing only the core of the pore complex with a minimal cytoplasmic density (17 nm in height, mini-pore, 73%, 4.6 Å), an extended pore (20 nm in height, extended-pore, 19%, 4.7 Å) and a full-length pore with a tall cytoplasmic region (27 nm in height, full-length-pore, 8%, 6.2 Å), respectively (Extended Data Figs. 2f–h and 4a–c). To better resolve the overall cytoplasmic regions, we merged the extended and full-length pore populations and obtained an overall resolution of 4.9 Å (referred to as full-pore hereafter; Fig. 1c and Extended Data Figs. 2l and 4d); the cytoplasmic crown including the prong was better resolved after focused classification and refinement (crown, 7.2 Å; Extended Data Figs. 2j and 4d). To improve the resolution of the core of the nsp3–nsp4 pore complex, we merged all populations and carried out a consensus refinement to reach an overall resolution of 4.2 Å (consensus-pore; Fig. 1b and Extended Data Fig. 2k), with a local resolution ranging from 3.9 Å in the centre to 7.1 Å at the peripheral regions (Extended Data Fig. 4e).
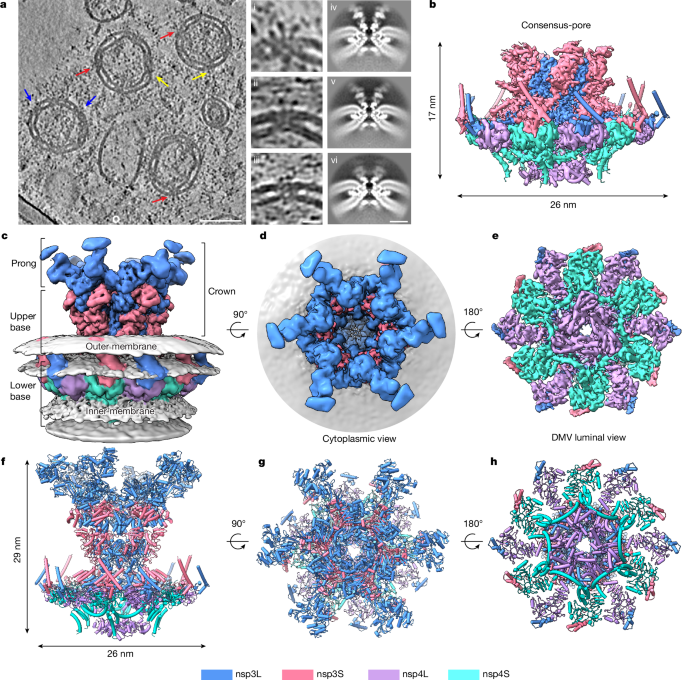
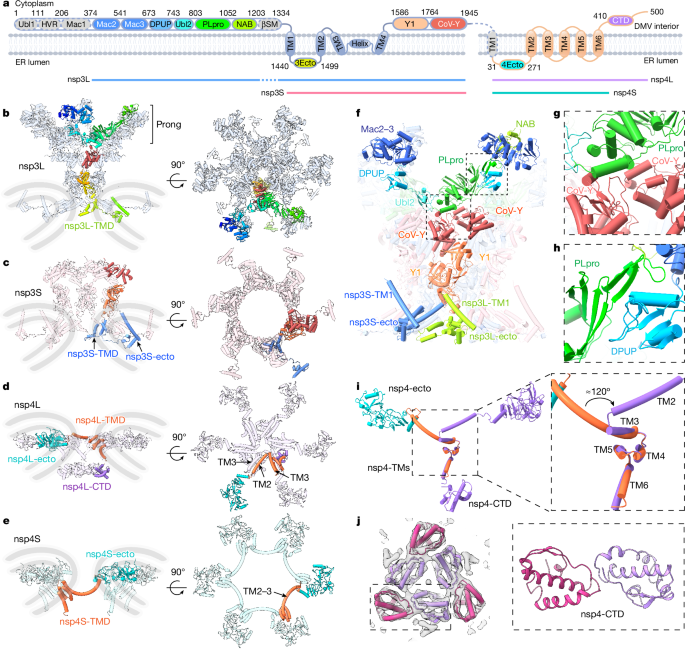
The local density of the central transmembrane (TM) region is sufficient for de novo model building, as exemplified in the TM helix (TMH) densities (Extended Data Figs. 5 and 6). nsp3 is the largest multifunctional protein encoded in the coronavirus genome (about 220 kDa)16, comprising ubiquitin-like domain 1 (Ubl1), a hypervariable region, macrodomains 1–3 (Mac1–3), a domain preceding Ubl2 and PLpro (DPUP), Ubl2, PLpro, a nucleic acid-binding (NAB) domain, a β-coronavirus-specific marker (βSM) domain, a TM domain (TMD), an ectodomain, Y1 and CoV-Y (Fig. 2a). nsp4 is a multipass TM protein without any known homologue. Using Alphfold217 and X-ray crystal structures of individual domains and homologues of nsp3, we built a near-complete structure of the nsp3–nsp4 DMV pore complex. Unexpectedly, the predicted carboxy-terminal domain (CTD) of nsp4 could not fit into the six-fold-symmetry density map in the DMV lumen, which was suspected to be due to symmetry mismatch. To resolve this region, we carried out symmetry relaxation and focused classification, revealing a three-fold symmetry of the nsp4-CTD dimer, instead of a six-fold symmetry (Fig. 1e,h, luminal view, and Extended Data Figs. 3 and 4f). Depending on the completeness of molecules that are resolved in the density map, we denote nsp3 and nsp4 as nsp3L (long form) and nsp3S (short form), and nsp4L (full-length) and nsp4S (short form with the CTD not visible), respectively (Figs. 1 and 2a). The complete nsp3–nsp4 pore complex is composed of 12 nsp3 and 12 nsp4 molecules (nsp3L/nsp3S/nsp4L/nsp4S = 6:6:6:6), with an overall six-fold symmetry except the nsp4-CTD (Fig. 1).
Topology and domain organization
From the top to bottom, we describe the DMV full-pore complex in three main parts: the prong, the upper base and the lower base (Fig. 1c). nsp3 is localized in the upper membrane to constitute the prong and upper base, whereas nsp4 sits in the membrane junction and bottom membrane to form the lower base (Fig. 2b–e). Most nsp3 domains (Mac2-Mac3-DPUP-Ubl2-PLpro-NAB, Y1-CoV-Y) are localized in the cytoplasmic side of the pore, constituting the DMV cytoplasmic ring, the diameter of which is substantially smaller than its counterparts in the spherules of alphavirus and nodavirus viruses8,18 (9 nm versus 15.6 nm and 19 nm, respectively; Extended Data Fig. 7a). The very N-terminal Ubl1-Mac1 of nsp3 is not visible in the full-pore, indicating their non-essential role in pore assembly, consistent with the recent finding in situ: ΔUbl1-Mac1 does not affect the overall architecture of the nsp3–nsp4 pore complex13 (Extended Data Fig. 7b). The rest of the N-terminal region of nsp3L (Mac2-Mac3-DPUP-Ubl2-PLpro-NAB), which constitutes the prong of the full-pore complex, lies above the C-terminal CoV-Y domain (Fig. 2b,f). The main interaction between the prong and the upper base is attributed to the interface formed by PLpro and CoV-Y (Fig. 2g). To accommodate this interaction with the CoV-Y base, Ubl2-PLpro adopts a bend of about 45° in the nsp3–nsp4 pore complex in contrast to the linearly aligned conformation observed for the isolated tandem domains in X-ray crystallography (Protein Data Bank (PDB): 4RNA and 6WRH; Extended Data Fig. 7c). The interaction within the prong seems to be stabilized by the interface of DPUP with the C terminus of PLpro from two protomers (Fig. 2h). These interfaces suggest an unexpected structural role of the PLpro domain in efficient pore formation, in agreement with the recent study in situ: ΔUbl1-Ubl2, which deletes the DPUP domain, affects the integrity of pores, but retains the capacity to form a crown-free pore complex13 (Extended Data Fig. 7b).
The C-terminal domains of nsp3 (TMD, nsp3-ecto, Y1-CoV-Y) form two concentric layers of hexamer, constituting the upper base underlying the prong in the cytoplasmic side (Fig. 2b,c). These two hexamers are anchored to the membrane by their TMD, which is revealed as comprising four TMHs (TM1–4) and a short horizontal helix (Fig. 2f and Extended Data Fig. 7d), instead of the previously predicted two helices. nsp3-TM1 is threaded by the N-terminus of nsp3-ecto, away from the central pore. TM2–4 of nsp3 are involved in scaffolding the central pore complex: TM3 and the horizontal helix are short helices buried inside the phospholipid bilayer whereas TM2 and TM4 connect with the ectodomain and the cytoplasmic Y1 domain, respectively (Fig. 2a). Depending on the position of nsp3 in the pore complex, the nsp3-TMDs vary slightly, specifically on the helix–TM4 hinge (root mean square deviation = 2.99 Å; Extended Data Fig. 7d). The angle of the Y1 and CoV-Y domains also differs by about 80° between the two nsp3 protomers (Extended Data Fig. 7e). The TMD and cytoplasmic Y1 and CoV-Y domains (hereafter referred to as nsp3C) are conserved among viruses in the order Nidovirales, whereas the N-terminal regions of nsp3 vary substantially19. Indeed, the N-terminal domains (Ubl1-NAB) of nsp3 are not visible in the mini-pore and extended-pore complexes (Extended Data Fig. 2f,g), in agreement with the fact that co-expression of nsp3C and nsp4 is sufficient to induce DMV formation12,20. The mini-pore complex further lacks the coronavirus-specific CoV-Y domain (Extended Data Fig. 2f), indicating that the TMD-ecto-Y1 domains19 act as the minimal requirement of nsp3 to constitute the pore complex in the order Nidovirales.
nsp4 contains six TMHs with its ectodomain (nsp4-ecto) localized in the intermembrane space and its CTD in the DMV lumen, forming two layers of concentric stacking hexamers constituting the lower base of the pore complex (Fig. 2d,e). The molecular interaction within the central interconnected nsp4 hexamer (nsp4L) is mainly mediated by two intermolecular antiparallel TMHs (TM2 and TM3), creating the central pore in the membrane junction (Fig. 2d). The other nsp4 hexamer (nsp4S) is localized at the bottom periphery of the pore whereas its ectodomains remain in the same horizontal position as the other hexamer ectodomains (Fig. 2e). Two nsp4 molecules adopt distinct conformations to accommodate its local environment in the pore complex: alignment of nsp4-TMDs reveals a rotation of about 120° of the nsp4-etco domain, which is mainly mediated by splitting of TM2 and TM3 in nsp4L, in sharp contrast with the continuously long curved TM2–3 helix in nsp4S (Fig. 2i). nsp4-CTD exhibits a trimer of dimer conformation, instead of a six-fold-symmetric hexamer (Fig. 2j and Extended Data Fig. 3). The nsp4-CTD trimer resembles the crystal structure of feline coronavirus (FCoV) nsp421, although FcoV-nsp4 appears to be twisted pentamers (incomplete hexamer) instead of flat trimer of dimer (PDB: 3GZF; Extended Data Fig. 7f). Overall, our structure of nsp3–nsp4 pore complex resolves the topology and domain organization of nsp3 and nsp4 in this unique pore complex.
Molecular contacts for nsp3–nsp4 pore formation
The ectodomain and TMD of nsp3 and nsp4 in the intermembrane space (Fig. 3a,b) and double-membrane junction (Fig. 3d,e) form the base of the pore complex. Twelve pairs of ectodomains of nsp3 and nsp4 form extensive contacts in the intermembrane space with a pseudo-12-fold symmetry, belting the pore complex from the intermembrane space (Fig. 3a). nsp3-ecto (V1458–Q1490) is predicted to be a disordered domain containing two pairs of conserved disulfide bonds. Our structure clearly defines the interface between the ectodomains of nsp3 and nsp4: nsp3-ecto attaches to the upper surface of nsp4-ecto, which is mediated by a combination of hydrophobic and hydrophilic interactions: V1458, D1478, L1480, Y1483, L1486 and Q1490 of nsp3 interact with nsp4 (Fig. 3c); the last four amino acid residues are more conserved (Fig. 3h), indicating their important roles in mediating nsp3–nsp4 interaction. Neutralization substitution of these hydrophobic residues (V1458A/L1480A) does not affect the interaction between nsp3 and nsp4, whereas a negatively charged mutant (V1458E/L1480E) reduces the interaction substantially. Alteration of the nsp3–nsp4 ectodomain interface by charge reversal (D1478E/Y1483E/L1486E/Q1490E) and charge neutralization (D1478A/Y1483A/L1486A/Q1490A) completely abolishes the interactions (Fig. 3i). Notably, nsp4 seems to be unstable in the absence of nsp3 interaction, as indicated by the presence of double bands and reduced protein levels. To determine whether these negatively charged substitutions affect DMV formation, we conducted in situ cryo-ET of transfected VeroE6 cells following cryo-focused ion beam milling. The double mutant (V1458E/L1480E) still revealed clustered DMVs and a characteristic feature of the pore complex (connection between two membranes in DMV) similar to the wild type, whereas the quadruple mutant (D1478E/Y1483E/L1486E/Q1490E) exhibited only multi-membrane vesicles and scarce DMV-like vesicles (Extended Data Fig. 8a,b). These data suggest that charge reversal at the nsp3–nsp4 ectodomain interface affects the capacity of DMV formation, although a larger cellular tomography dataset is required to statistically quantify the effect.
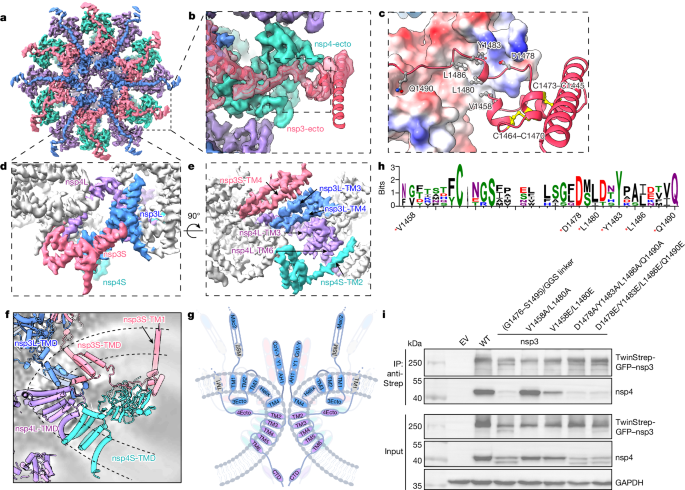
The TM region is formed by 120 TMHs from 12 copies each of nsp3 and nsp4, stacked into four concentric layers (Fig. 3d–g). nsp3S-TM4 and nsp3L-TM3 mediate the interactions between the top two layers of nsp3-TMDs. The central nsp4L links the middle nsp3L layer and the bottom nsp4S layer by nsp4L-TM3 and nsp4L-TM6, respectively (Fig. 3d,e). A longitudinal cross-section of the TM regions reveals multiple interlocked interactions between TMDs to create a sharp membrane curvature (87°; Extended Data Fig. 7a), which is tipped by the turning point between two TMHs in nsp4L (TM2 and TM3; Fig. 3f,g). The membrane curvature within the DMV pore is larger than that in membrane spherule necks induced by alphavirus and nodavirus (87° in coronavirus versus 139° in alphavirus and 111° in nodavirus, respectively; Extended Data Fig. 7a). The curvature of the DMV pore membrane seems to be stabilized by the interaction of ectodomains in the intermembrane space, with nsp3-TM1 anchored into the outer membrane away from the central pore (Fig. 3f,g). Additionally, the bottom of nsp3-Y1 is highly positively charged, complementary to the negatively charged phospholipid head group in the concave upper DMV membrane (Extended Data Fig. 9a), which may also contribute to the optimal stability of the nsp3–nsp4 pore complex. The topology and intermolecular interactions of nsp3 and nsp4 in the DMV pore suggest a possible membrane reorganization pathway to drive DMV pore formation from endoplasmic reticulum membrane: cleavage of nsp3 and nsp4 by nsp3-PLpro releases the spatial restraint between the nsp3-CTD and nsp4-ecto, allowing the nsp3-CTD (Y1 and CoV-Y domains) to oligomerize; meanwhile, the interaction of TMDs and cis interaction of nsp3–nsp4 ectodomains on the same membrane lead to the formation of the highly locally curved membrane for the pore to assemble (Extended Data Fig. 8c). This could further develop into a paired membrane sheet with double-membrane-spanning pores stabilizing it, as observed previously in situ13. However, how these paired membranes develop into closed DMVs remains unknown and experimental evidence is required to substantiate this hypothesis. By contrast, in the nsp3–nsp4-cleavage-deficient mutant, the nsp3-CTD is constrained close to the membrane, which may render nsp3 unable to oligomerize; the trans interactions of nsp3-ecto with nsp4-ecto between two membranes would lead to a membrane zipper13, instead of a membrane with a local high curvature (Extended Data Fig. 8c).
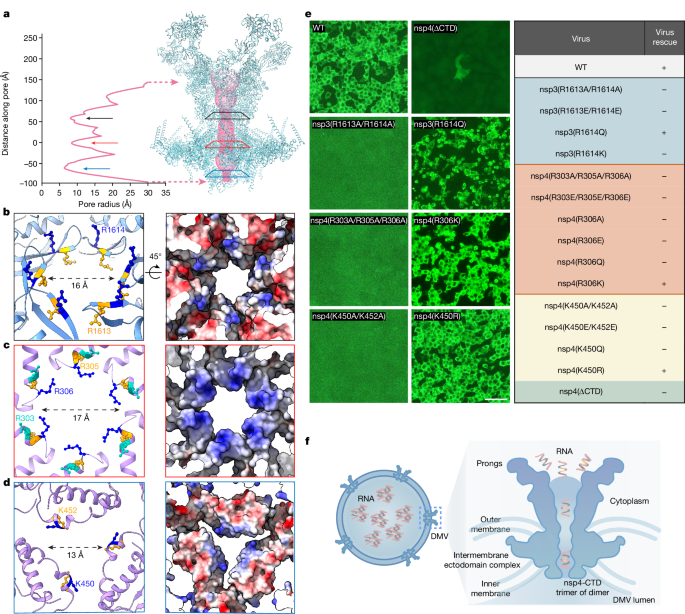
Arginine ring-coordinated DMV pore
The DMV pore complex is postulated to mediate RNA translocation and exhibits an overall 25-nm-long funnel-shaped channel with three constriction sites (Fig. 4a). The nsp3-Y1 domain hexamer forms the top constriction site (R1613/R1614; Fig. 4b), and the central pore is created by nsp4-TMDs (R303, R305 and R306 at the TM2–TM3 junction; Fig. 4c). The bottom pore is formed by the nsp4-CTD trimer of dimer (K450 and K452; Fig. 4d). These positively charged residues are highly conserved across β-coronaviruses (Fig. 4b–d and Extended Data Fig. 10a). Specifically, nsp4 R306 faces its side chain directly into the central pore, potentially gating the transport of nucleotide and newly synthesized viral RNA. The central pore has a diameter of 17 Å, which is sufficient for small-molecule metabolites and single-stranded RNA transport, but may be insufficient to accommodate double-stranded RNA (which is estimated to be 24 Å in diameter). Consistent with the putative function of RNA transport, the central pore of DMV exhibits an overall positively charged inner surface (Extended Data Fig. 9b). To probe the functional importance of these positively charged residues at the constriction sites in virus replication, we introduced site-directed mutagenesis in a SARS-CoV-2 reverse genetics system based on bacterial artificial chromosome (BAC)22. Both charge-neutralization (nsp3(R1613A/R1614A), nsp4(R303A/R305A/R306A) and nsp4(K450A/K452A)) and charge-reversal (nsp3(R1613E/R1614E), nsp4(R303E/R305E/R306E) and nsp4(K450E/K452E)) substitutions abolished the viral replication capacity (Fig. 4e). To explore whether the virus can tolerate milder substitutions, we introduced single amino acid residue substitutions at these three constriction sites. For the central pore and bottom pore, substitutions with positively charged residues (nsp4(R306K) and nsp4(K450R)) retained viral replication activity, whereas charge-neutralization (nsp4(R306A)) and charge-reversal (nsp4(R306Q) and nsp4(R306E)) substitutions failed to rescue the virus’s capacity of effective replication (Fig. 4e). Conversely, virus carrying nsp3(R1614Q) can be rescued whereas that carrying nsp3(R1614K) cannot. Notably, these substitutions did not affect the interaction between nsp3 and nsp4, and DMV formation (Extended Data Fig. 10b,c). We further examined the structure of neutralization mutants by subtomogram averaging. The substitutions in the central pore (nsp4(R303A/R305A/R306A)) did not affect the architecture of the nsp3–nsp4 pore complex, whereas the substitutions in the cytoplasmic crown (nsp3(R1613A/R1614A)) and luminal region (nsp4(K450A/K452A)) of the pore complex compromised the integrity of pores, as indicated by the blurring of the cytoplasmic crown and luminal regions in the two mutants, respectively (Extended Data Fig. 10d). Collectively, these data suggest a specific and critical role of these positively charged residues in virus replication, potentially in mediating metabolites and RNA transport.
Discussion
Intracellular membrane remodelling is a common feature of positive-stranded RNA virus replication23. The structural and functional analogy of the DMV pore complex to the replication complex of other positive-stranded RNA virus replication organelles, such as the replication complex formed by chikungunya virus8,24 and nodavirus7, indicates a broadly conserved mechanism for RNA replication and translocation. However, the viral genome synthesis and capping activities are integrated into the pore complexes of the spherules induced by alphavirus and nodavirus. By contrast, coronavirus RNA replication machinery is not an integral component of the DMV pore complex, although they might be transiently associated with each other3, which warrants further investigation. The DMV pore complex architecture broadly resembles a miniature nuclear pore complex (NPC): the cytoplasmic prong, which comprises large nsp3 N-terminal domains, is analogous to the NPC filaments for cargo recruitment, including host factor and RNA binding; the organization of the double-membrane compartment is similar to the NPC membrane in the nuclear envelope, and the intermembrane nsp3–nsp4 ectodomain complexes resemble the NPC luminal rings to maintain the integrity of pore complex; finally, the DMV nsp4-CTD trimer of dimer inside the DMV may resemble the NPC basket contributing to membrane shaping forces within the NPC and providing an anchoring site for RNA export25.
Our structures of the nsp3–nsp4 pore complex resolve the long-standing question regarding the stoichiometry and topology of nsp3 and nsp4 within the DMV pore, suggesting a potential pathway of membrane zippering and DMV pore formation. Additionally, the nsp3–nsp4 pore complex structure reveals the structural role of nsp3-NTD (Mac2-Mac3-DPUP-Ubl2-PLpro-NAB) in crowning the pore, whereas the conserved nsp3-CTDs (comprising 4 TMHs, nsp3-ecto, Y1 and CoV-Y) and nsp4 serve as the base of the pore complex. The three constriction sites in the nsp3–nsp4 pore complex provide an overall positively charged channel for RNA and metabolite transport. Considering the conservation of nsp3-CTD and nsp4 among coronaviruses and torovirus in the Coronaviridae family26, our structures establish a framework for understanding DMV pore formation and RNA translocation in the Coronaviridae family (Fig. 4f), which may even extend to viruses in the Arteviridae family of Nidovirale19,27.
The architecture of our nsp3–nsp4 full-pore complex closely resembles that of the native authentic DMV pore complex observed during MHV infection in situ3. The conformation of the nsp3–nsp4 complex without RNA may represent a resting state of the DMV pore during RNA and metabolite transportation. However, owing to the absence of RNA synthesis machinery in our minimal DMV system, our nsp3–nsp4 complex may not fully recapitulate the complete functional DMV pore complex during virus infection. We identified three main conformations of nsp3–nsp4 pore complex in our dataset, with the mini-pore complex as the prevalent species (Extended Data Fig. 2e). By contrast, the same complex in situ was found as a full-pore complex13, with a minor population of mini-pores. The heterogeneity of the nsp3–nsp4 complexes in vitro could be attributed to both conformational flexibility and sample degradation during DMV isolation, with the latter probably being the primary factor. Nonetheless, considering that the nsp3-NTD is stacked onto the upper base of the pore complex by interacting with the CoV-Y ring in the full-pore complex, it is also possible that the shorter forms (mini- and extended-pores) represent the intermediate states towards full-pore complex assembly.
Perturbing DMV pore formation and viral RNA translocation activity represents a new approach to viral inhibition. Our study will facilitate the rational design of new antiviral strategies against SARS-CoV-2 and other coronaviruses based on interference of DMV pore formation and inhibition of DMV pore function, expanding the current targets28,29. For example, interference of the inter-ectodomain interaction of nsp3 and nsp4 by nsp3-mimicking peptides would inhibit DMV pore formation; negatively charged molecules could be designed to target the central arginine pore, blocking RNA and metabolite transport. Our experimental approach highlights the great potential of cryo-ET and subtomogram averaging in studying in vitro systems recapitulating in situ environments30. The isolated coronavirus DMVs therefore provide a valuable in vitro system for investigation of the virus replication mechanism at near-atomic resolution, setting the foundation for dissecting the structure and function of the complete replication–transcription complex, which comprises a DMV pore complex and RNA replication–transcription machinery.
Methods
Plasmid constructs
Codon-optimized SARS-CoV-2 nsp3 and nsp4 sequences from the Wuhan-Hu-1 SARS-CoV-2 genome were amplified by the donor plasmids pEGFPN1-nsp3-EGFP and pmCherryN1-nsp4-mCherry, which were purchased from Addgene (plasmid numbers 165108 and 165132). The insertions nsp3 and nsp4 were assembled using overlap-extension PCR followed by insertion into a destination pcDNA3.1 vector containing an N-terminal TwinStrep-EGFP tag using the ClonExpress II One Step Cloning kit (Vazyme, C112) to produce pcDNA3.1-TwinStrep-EGFP-nsp3-nsp4. The presence of the EGFP tag allows the timely assessment of transfection efficiency and the subsequent DMV isolation procedure. Site-directed mutagenesis was introduced to the full-length construct by overlapping PCR. All of the constructs were confirmed by sequencing (BGI Genomics).
Cell culture and antibodies
Expi293F cells (Expi293 Expression System Kit, A14635) were purchased from Thermo Fisher Science and were cultured in OPM-293 CD05 medium (OPM Biosciences, 81075-001) supplemented with 100 U ml−1 penicillin and 100 U ml−1 streptomycin at 125 r.p.m., 37 °C and 5% CO2. Suspension cultures of HEK293F cells were grown to a density of about 3 × 106 cells per millilitre and transfected with plasmids using PEI MAX (Polysciences), which can achieve 60–70% transfection efficiency. At 18–20 h post-transfection, 2.5 mM sodium butyrate was added, and cell pellets were collected after about 72 h by centrifugation followed by washing with 1× phosphate-buffered saline (PBS). The cell pellets were frozen in liquid nitrogen and stored at −80 °C until further use. HEK293T cells and VeroE6 cells (Research Resource Identifier CVCL_0574) were cultured in Dulbecco’s modified Eagle medium (Thermo Fisher Science) supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin and 100 U ml−1 streptomycin at 37 °C and 5% CO2. HEK293T cells and VeroE6 cells were transfected with plasmids using Lipofectamine 3000 (Thermo Fisher Science, L3000015) according to the manufacturer’s instructions.
Co-immunoprecipitation and western blot analysis
HEK293T cells were transfected with the wild-type or mutant pcDNA3.1-TwinStrep-EGFP-nsp3-nsp4 plasmids. At 48 h post-transfection, HEK293T cells were washed with 1× PBS and lysed using ice-cold cell lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% glycerol and 1% Triton X-100, supplemented with protease inhibitor cocktail (Sigma-Aldrich, P8340) and GENIUS Nuclease (ACROBiosystems, BEE-N3116)). After incubation on ice for 30 min, cell debris was cleared by centrifugation at 12,000 r.p.m. for 30 min at 4 °C. The supernatants were incubated with Strep-TactinXT 4Flow resin (IBA LifeSciences, 2-5010-025) for 3 h at 4 °C. The resin was then washed three times with ice-cold lysis buffer, and the bound proteins were eluted with 2× SDS sample buffer (0.2 M Tris-HCl (pH 6.5), 0.4 M dithiothreitol, 8% SDS, 6 mM bromophenol blue and 4.3 mM glycerol). Samples were then subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and western blot analysis. Proteins were separated by SDS–PAGE and transferred to a polyvinylidene difluoride membrane (Millipore, IPVH00010). The membranes were subsequently blocked with 5% nonfat milk (Santa Cruz, sc-2325) in TBST (50 mM Tris-HCl (pH 7.4), 150 mM NaCl and 0.1% Tween-20) for 2 h. nsp3 and nsp4 were probed using the primary and secondary antibodies indicated below, and developed with a chemiluminescent substrate (Thermo Fisher Science, 34095). Protein bands were visualized on the Bio-Rad ChemiDoc MP Imaging System (Bio-Rad). The following antibodies were used for western blots: rabbit polyclonal anti-SARS-CoV-2 nsp3 (Thermo Fisher Science, PA5-116947, dilution 1:5,000), rabbit polyclonal anti-SARS-CoV-2 nsp4 (Abclonal, A20281, dilution 1:1,000) and mouse monoclonal anti-GAPDH (Santa Cruz, sc-47724, dilution 1:5,000) were used as the primary antibodies. Anti-rabbit IgG–HRP antibody (Cell Signaling, 7074S, dilution 1:2,000) and anti-mouse IgG–HRP antibody (Cell Signaling, 7076S, dilution 1:2,000) were used as secondary antibodies.
SARS-CoV-2 nsp3–nsp4 DMV purification
Frozen HEK Expi293F cell pellets from 1 l cell culture were suspended in 80 ml hypotonic buffer (20 mM HEPES (pH 7.5), 1.5 mM MgCl2 and 10 mM KCl) with protease inhibitor cocktail (Sigma-Aldrich, P8340) and GENIUS nuclease (ACROBiosystems, BEE-N3116). Cells were homogenized with a 50-ml Dounce homogenizer (Thomas Scientific) by repeated plunging for 40 strokes. The homogenate was then transferred to a beaker and sonicated in an ice bath with a probe sonicator (Branson Digital Sonifier SFX 550). The homogenate was disrupted for 15 cycles (2 s on and 4 s off) at 30% power. A total of 60 cycles were carried out with a 30-s delay between each 15 cycles. The homogenate was spun at 4,000g for 20 min to remove the cell debris and the resulting supernatant was loaded to 2.5 ml Strep-TactinXT 4Flow resin (IBA LifeSciences, 2-5010-025) pre-equilibrated with an equilibrate buffer (50 mM HEPES (pH 7.5), 150 mM NaCl and 1 mM EDTA) for binding for 3 h in a gravity column. The resin was further washed with 10 column volumes of washing buffer (50 mM HEPES (pH 7.5), 500 mM NaCl and 1 mM EDTA) and then 10 column volumes of equilibrate buffer. The vesicles were eluted by elution buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA and 50 mM biotin). For each elution step, the Strep-Tactin resin was incubated with elution buffer for 15 min. All elution fractions were pooled together and loaded into a 13-ml ultracentrifugation tube and spun at 200,000g in a Thermo Scientific Sorvall WX ultracentrifuge with a TH-641 swing-bucket rotor for 1 h. The final pellet was gently suspended in STE buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl and 1 mM EDTA). All steps were finished within 1 day and samples were kept on ice or 4 °C throughout the purification procedure. Cryo-EM grids were prepared immediately after DMV resuspension.
Cryo-EM grid preparation for isolated DMVs
The 6-nm bovine serum albumin-coated gold fiducial beads (Aurion, 206.033) were concentrated 10 times by benchtop centrifugation for 30 min at 4 °C. The gold beads were added to samples before freezing. A 3.5 µl volume of purified DMV samples was applied to freshly glow-discharged lacey carbon grids (Agar Scientific, AGS166-3, 300 mesh). The grids were blotted using a Vitrobot Mark IV with a blot force of 0 and blot time of 3.5 s with 95% humidity at 4 °C, and flash-frozen in liquid ethane before being stored in liquid nitrogen until data collection. Cryo-EM grids were screened in a 200-kV Thermo Scientific Glacios microscope with a Falcon 4 camera to optimize freezing conditions.
Cryo-ET data collection
The tilt series were acquired using a Thermo Fisher Krios equipped with a Falcon 4i camera and Selectris energy filter, with a slit width of 20 eV. PACE-tomo script31 within serialEM32 was used and a maximum of 15-μm image–beam shift was allowed when adding acquisition points. For purified DMV samples, a dose-symmetric scheme (group of 2) was used, with a tilt range of −51° to 51° (or −60° to 60°) at 3° increments and an exposure dose of 3 electrons per square ångström per image at a magnification of ×81,000 (pixel size: 1.571 Å). Tilt series were acquired with a defocus between −1 µm and −6 µm and a total of 5,170 tilt series were acquired. The tilt-series data were collected using multiple grids from two independent DMV purifications over multiple data collection sessions on the same microscope. The detailed data collection parameters are listed in Extended Data Table 1.
Subtomogram averaging and classification
The raw micrographs in EER format were motion-corrected using relion_motioncor, applying gain reference correction. The blank images were removed by calculating the average image intensity using the clip command in IMOD. The motion-corrected images were then stacked into individual tilt series and aligned using batchruntomo, using the previous Python script (tomo_toolbox.py: https://github.com/ffyr2w/cet_toolbox). The resulting tilt series were imported to emClarity (v1.6.215) for contrast transfer function estimation, followed by particle picking with 8×-binned tomograms for template-matching (EMD-11514). The coordinates of picked particles and contrast transfer function information were exported to RELION (v4)33 for further 3D classification and 3D refinement. The initial 3D classification with C6 symmetry was carried out with 4×-binned pseudosubtomograms to remove the incorrectly picked particles. A total of 113,163 particles were obtained from the template-picked particles (598,699) and pulled together for another round of 3D classification into 6 classes. On average, about five nsp3–nsp4 complexes can be identified from each DMV after 3D classification, which is estimated from a subset of dataset containing 5,101 particles from 996 DMVs. The six classes were pooled into three main classes depending on the appearance of the cytoplasmic density of the pore complex and refined separately. The refinement was carried out sequentially with 4×-, 2×- and 1×-binned pseudosubtomograms with C6 symmetry, with three rounds of tomo frame alignment in total, which resulted in density maps of a mini-pore at 4.6 Å, an extended-pore at 4.7 Å and a full-length-pore at 6.2 Å. Two classes (extended-pore and full-length-pore) were merged together to improve the cytoplasmic density, achieving a final resolution of the consensus full-length pore complex (full-pore) at 4.9 Å. To improve the resolution of the core region of the pore complex, all of the particles were pooled together and refined as above, reaching a final resolution of 4.2 Å in the consensus map (consensus-pore). Focused classification and refinement of the cytoplasmic crown in the full-length-pore was carried out to improve the density of the crown. Particle centres were shifted using relion_star_handler during focused classification and refinement. The subtomogram averaging and classification pipeline is summarized in Extended Data Fig. 2.
To resolve the density in the DMV lumen of the pore complex, we carried out focused classification of this region (skipping alignment) with either C3 or C1 symmetry, which resulted in classes with clear features of the nsp4-CTD, forming a trimer of nsp4-CTD dimer (Extended Data Fig. 3). The classes with discernible density of nsp4-CTD trimer of dimer were pooled together, and two classes were rotated by 180° along the z axis to merge into the other classes. The final map was further refined and reconstructed with C3 symmetry and sharpened with default b-factors, as determined by a Guinier plot. The local resolution was estimated in RELION (Extended Data Fig. 4).
Model building and validation
We first built the model using the C6-symmetry consensus-pore map at an overall 4.2 Å resolution. The individual domains of nsp3 and nsp4 were predicted by Alphafold217 and cross-validated with the X-ray crystal structures (Supplementary Figs. 2 and 3). The ectodomains of the nsp3–nsp4 complex were predicted as a complex (Supplementary Fig. 4) and were placed into the density map manually together with other individual domains in Chimera. The predicted TMDs were manually fitted into the density maps, followed by manual real-space refinement in Coot34. The resulting structure was further refined by ISOLDE35 implemented in ChimeraX36. The final refinement enabling only rigid-body refinement and atomic displacement parameter (b-factor) refinement was conducted by phenix_refine37. The sequence register is validated by many bulky hydrophobic residues in the TMD (Extended Data Fig. 5c), and further checked using the checkMySequence tool38. The asymmetric unit of the consensus-pore complex contains four chains as follows—nsp3L: 1411–1945 and nsp3S: 1403–1945; nsp4L and nsp4S: 31–401.
To model the C3-symmetry consensus-pore complex, the asymmetric unit refined from the C6-symmetry consensus-pore complex and Alphafold-predicted nsp4-CTD was manually fitted into the map. The structures were refined in ISOLDE and phenix_refine as above. To model the full-pore complex, we used the asymmetric unit from the C6-symmetry consensus-pore complex as the starting model and manually fitted the Alphafold-predicted DPUP-Ubl2-PLpro and CoV-Y domains into the map. The prong tip is in low resolution (12–15 Å resolution) and the Mac2–3 and NAB domain were predicted as a complex using AlphaFold2 (Supplementary Fig. 5) to rigid-body fit into the map. The unresolved region between NAB and nsp3-TM1 consists of >200 amino acid residues, which is predicted to contain two helices, a βSM domain and a long-disordered loop (Supplementary Fig. 2d). The disordered loop (residues 1199–1241) can span a distance of 14 nm, and with the additional βSM domain (which is 4 nm in length), this unresolved region is sufficiently long to link NAB and nsp3L-TM1. The final structures were refined in ISOLDE and phenix_refine as above. Residues showing high clashing scores in phenix were manually corrected in Coot. The regions that are modelled in the full-pore map include the following: nsp3L: 417–1945; nsp3S: 1403–1945; nsp4L: 31–500; and nsp4S: 31–401. Most of the domains generated by Alphafold are similar to the final refined models except the TMDs. The statistics of Alphafold starting models, per-residue Q-scores using MapQ39 and overfitting assessment40 are presented in Extended Data Fig. 6 and Supplementary Figs. 6 and 7. The local-resolution-filtered maps are used for presentation in Chimera and ChimeraX. The structure alignment and comparison are carried out in Chimera using the matchmaker command.
Flow cytometry of transfected VeroE6 cells and cryo-EM grids preparation
VeroE6 cells were transfected with the wild-type or mutant pcDNA3.1-TwinStrep-EGFP-nsp3-nsp4 plasmids. At 24 h post-transfection, VeroE6 cells were washed with 1× PBS and treated with 0.25% trypsin–EDTA solution at 37 °C until cells were detached from the bottom of the 10-cm culture dish. Cells were pelleted at 200g for 3 min and washed with 1× PBS, and then resuspended in 500 μl FACS buffer (1× PBS, 25 mM HEPES (pH 7.5), 2% fetal bovine serum, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin). The cell suspensions were filtered through 40-μm Falcon Cell Strainers (BD, 352340) before acquisition on a flow cytometer. Single-cell sorting and FACS analysis were carried out using a BD FACSAria SORP Cell Sorter with a 488-nm laser. Cells were considered positive when the fluorescence intensity was above a threshold value that was determined by the maximum intensity of the non-transfected control cells. Cells with GFP fluorescence signal were sorted and then seeded on the carbon side of the freshly prepared EM grids (Quantifoil Gold R2/2, 200 and 300 mesh) in 6-well plates. The grids were pre-treated as follows: glow-discharged for 45 s at 15 mA (PELCO easiGlow) and transferred, carbon side up, to a 6-well plate, then treated with 20 µg ml−1 of bovine plasma fibronectin (Sigma) in PBS for 30 min and washed with PBS three times. Then the grids were UV-treated for 1 h. After the cells were seeded, the 6-well plates were incubated at 37 °C with 5% CO2 until plunge-freezing.
Plunge-freezing of cells grown on the EM grid
Grids were picked up from the 6-well plates and loaded into a Leica GP2 plunge freezer. The ethane temperature was set to −184 °C and the chamber was set to 37 °C with 80% humidity. An additional 3.5 μl of cell culture medium was applied to the back side of the grids, which were then blotted from the back for 7 s. The grids were immediately plunge-frozen in liquid ethane after blotting. Vitrified grids were stored in liquid nitrogen for further processing.
Cellular lamella preparation, cryo-ET data collection and tomogram reconstruction
Grids were clipped into cryogenic focused ion beam AutoGrids (Thermo Fisher Scientific). Lamella preparation was carried out on an Aquilos 2 cryogenic focused ion beam system (Thermo Fisher Scientific) using AutoTEM software. Grids were sputter-coated with platinum for 30 s, followed by 20 s of gas injection system coating. The gallium ion beam was gradually adjusted to lower values as the lamella thinning progressed (0.5 nA until 3 µm thick, 0.3 nA until 1.5 µm, 0.1 nA until 0.75 µm, then 50 pA until 300 nm and finally 30 pA until about 150–200 nm thickness).
The tilt series were acquired as videos on a Thermo Fisher Krios microscope operated at 300 kV with a Selectris energy filter using serialEM. The pre-tilt angle was estimated by comparing the medium-mag images of lamella acquired at ±45°. The tilt series were collected with a tilt range of −60° to 48° at 2° increments, a target defocus of −15 µm and an exposure dose of 1 electron per square ångström per image at a magnification of ×15,000 (pixel size: 8.571 Å). The raw micrographs in EER format were motion-corrected using relion_motioncor with gain reference applied. Dark images due to high tilt or neighbour ice contamination were removed by calculating the average image intensity using the clip command in IMOD. The motion-corrected images were then stacked into individual tilt series and aligned using AreTomo41. Tomograms were denoised using Topaz42 for better visualization.
Generation of recombinant SARS-CoV-2 with alterations
The infectious clone of SARS-CoV-2 on a BAC, named p-BAC-SARS-CoV-2, was generated and characterized as described previously22,43. The recombinant SARS-CoV-2 with nsp3 and nsp4 alterations was generated using homologous recombination. Briefly, two guide RNAs, sgRNA1 and sgRNA2 (Sangon), were used for CRISPR–Cas9 cleavage of p-BAC-SARS-CoV-2 with the Cas9 enzyme digestion kit (NEB) according to the manufacturer’s instructions. The linearized p-BAC-SARS-CoV-2 was verified by gel electrophoresis and purified with a Gel Extraction Kit (Qiagen). The site-directed mutagenesis of nsp3 and nsp4 was carried out by overlapping PCR with primers F and R. The resulting gene fragments were further inserted into the linearized p-BAC-SARS-CoV-2 through homologous recombination using ClonExpress II One Step Cloning Kit (Vazyme). The primers are shown in Extended Data Table 2.
Recovery of recombinant viruses and immunofluorescence assay
BHK21-ACE244 cells at about 80% confluence in a six-well culture plate were transfected with 3 μg recombinant p-BAC-SARS-CoV-2 using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. At 6 h post-transfection, the cells were trypsinized and added as an overlay to infect VeroE6-TMPRSS244 cells in a six-well plate for 72 h. Cells were observed daily for the appearance of cytopathic effects. Also, the cell culture supernatant was collected at 24, 48 and 72 h post-transfection for detection of live virus titre and genome copies. A mutant virus without a steady increase of virus titre or copies was deemed as failed to be rescued.
Immunofluorescence assay was carried out to verify the rescue of recombinant virus. Cells were fixed with 4% paraformaldehyde in PBS at room temperature for 30 min, and then the cell membrane was permeabilized with 0.2% (vol/vol) Triton X-100 in PBS for 5 min. Cells were blocked in 4% bovine serum albumin buffer for 30 min at 37 °C, washed with PBS, and incubated with the primary antibody (SARS-CoV-2 N polyclonal antibody45, 1:4,000) overnight at 4 °C, followed by washing with PBST for three times. The cells were then stained with Alexa Fluor 488 goat anti-rabbit IgG (H + L; Thermo Scientific, A-11064, 1:1,000) for 1 h at room temperature. After being washed with PBST, cells were visualized and imaged under a fluorescence microscope (Olympus).
Mass spectrometry
Protein samples were resolved using SDS–PAGE gel and visualized with Coomassie stain. The protein bands were cut into separate slices and subjected to in-gel digestion. Briefly, gel slices were subjected to reduction and alkylation by 10 mM TCEP and 55 mM 2-chloroacetamide, respectively. Protein digestion was carried out by incubating with trypsin (1 ng µl−1) overnight at 37 °C. Subsequent tryptic peptides were extracted from the gel with 50% ACN/5% FA and 100% ACN sequentially. The peptide extracts were pooled together and SpeedVac dried. The peptides were desalted using C18 StageTips for analysis by liquid chromatography with tandem MS spectrometry (MS) analysis. Eluted peptides were analysed with a nanoelute UHPLC coupled to a Bruker timsTOF pro mass spectrometer. The peptide mixture was loaded onto an Aurora C18 UHPLC column (75 μm i.d. × 25 cm length × 1.6 μm particle size (IonOpticks)). Chromatographic separation was carried out using a linear gradient of 2–30% of buffer B (0.1% FA in ACN) at a flow rate of 300 nl min−1 over 27 min. MS data were collected over an m/z range of 100 to 1,700. During MS/MS data collection, each thermal Ionization MS cycle was 1.1 s and included 1 MS plus an average of 10 PASEF MS/MS scans. Raw mass spectrometry data were processed using MaxQuant 1.6.14.0. Raw data were searched against the SARS-CoV-2 FASTA database containing 17 entries and the Human Swissprot FASTA database containing 20,361 entries, using the following settings: oxidized methionine and acetylation were selected as dynamic modifications, and carbamidomethyl as a fixed modification with a minimum peptide length of 7 amino acids was enabled. Confident proteins were identified using a target–decoy approach with a reversed database, a strict false-discovery rate of 1% for the peptide and peptide spectrum match level, and a minimum of ≥1 unique peptide and ≥2 peptide spectral matches.
Sequence conservation and pore radius analysis
The protein sequences were analysed using SnapGene Viewer (v7.1.1). The conservation plot was generated with WebLogo46 (https://weblogo.threeplusone.com/) using the protein sequences shown in Supplementary Fig. 9. HOLE was used for calculating the nsp3–nsp4 pore radius and central channel dimensions47.